Wat is het Angelman syndroom?
Het Angelman syndroom is een syndroom waarbij kinderen een ontwikkelingsachterstand hebben vaak in combinatie met ernstige spraaktaalproblemen, een kenmerkend uiterlijk, epilepsie en slaapproblemen als gevolg van een verandering in het erfelijk materiaal van chromosoom 15.
Hoe wordt het Angelman syndroom ook wel genoemd?
Het Angelman syndroom is genoemd naar een kinderarts Angelman die dit syndroom beschreven heeft. Het Angelman syndroom wordt wel afgekort met de letters AS.
Happy puppet syndroom
Kinderen met het Angelman syndroom zijn vaak vrolijk, lachen veel en wapperen met hun handen. Dit deed mensen denken aan het gedrag van een blije puppy. Vroeger werd daarom de naam Happy puppet syndroom wel gebruikt, maar tegenwoordig wordt deze naam steeds minder gebruikt, omdat het Angelman syndroom een ernstige aandoening is die grote gevolgen heeft voor de ontwikkeling van een kind.
Prader-Willi syndroom
Het Angelman syndroom is verwant aan het Prader-Willi syndroom. Bij dit syndroom mist hetzelfde stukje erfelijk materiaal, maar dan mist juist het stukje erfelijk materiaal van de vader en niet van de moeder.
15q duplicatie syndroom
Het Angelman syndroom is ook familie van het 15q duplicatiesyndroom. Kinderen met het Angelman syndroom missen een stukje van het erfelijk materiaal van chromosoom 15, terwijl kinderen met het 15q duplicatie syndroom juist te veel aan chromosoom 15 hebben.
Hoe vaak komt het Angelman syndroom voor?
Het Angelman syndroom is een zeldzame ziekte. Het Angelman syndroom komt ongeveer bij één op de 10.000 tot 25.000 kinderen voor. In Nederland worden elk jaar gemiddeld tussen de 10 en 20 kinderen geboren met het Angelman syndroom.

Bij wie komt het Angelman syndroom voor?
Het Angelman syndroom is al vanaf de geboorte aanwezig. Vaak wordt tijdens het eerste levensjaar duidelijk dat de ontwikkeling van het kind anders verloopt dan die van andere kinderen.
Het Angelman syndroom komt even vaak bij jongens als bij meisjes voor.
Waar wordt het Angelman syndroom door veroorzaakt?
Fout in erfelijk materiaal
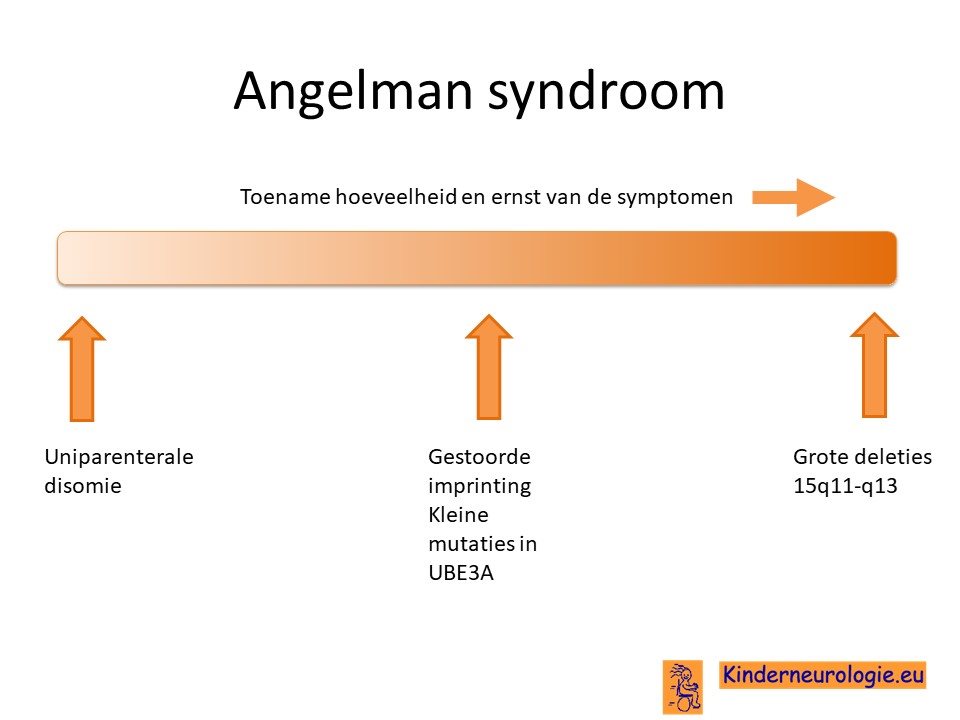
Het Angelman syndroom wordt veroorzaakt door een foutje op een stukje materiaal op het 15e-chromosoom, om precies te zijn op 15q11.2 van het 15e chromosoom. Het blijkt dat kinderen dit stukje 15q11.2 alleen van de vader hebben gekregen en niet van de moeder. Er zijn verschillende manieren mogelijk die er voor zorgen dat kinderen dit stukje van chromosoom 15 alleen van de vader krijgen.
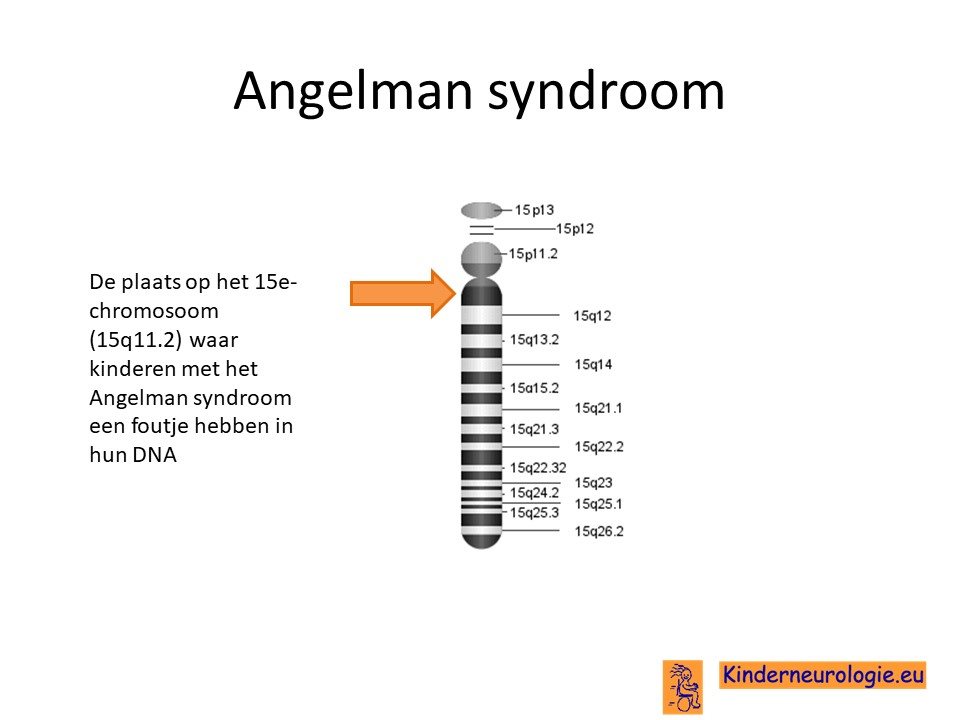
Ontbreken deel moederlijke deel chromosoom 15
Bij zeven van de tien kinderen ontstaat het Angelman syndroom omdat er een stukje van chromosoom 15 mist van het chromosoom15 wat het kind van de moeder heeft gekregen. Het missen van en stukje van het erfelijk materiaal wordt een deletie genoemd. Er wordt onderscheid gemaakt in 2 soorten deleties: deletie type 1 en deletie type 2. Deletie type 1 is groter dan deletie type 2. Kinderen met een type 1 deletie hebben meestal meer problemen dan kinderen met een type 2 deletie. Kinderen met een deletie hebben in het algemeen weer meer problemen dan kinderen met een uniparenterale disomie.

Fout in het UBE3A-gen
Bij een op de vier kinderen met het Angelman syndroom is er sprake van een fout in een deel van chromosoom 15 wat het UBE3A-gen wordt genoemd.
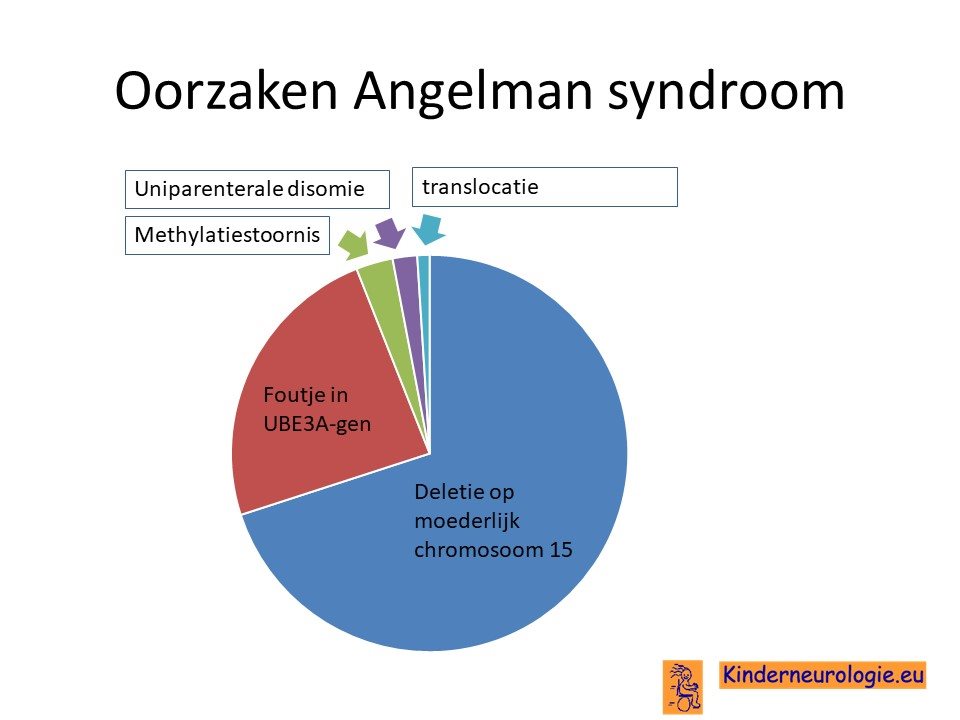
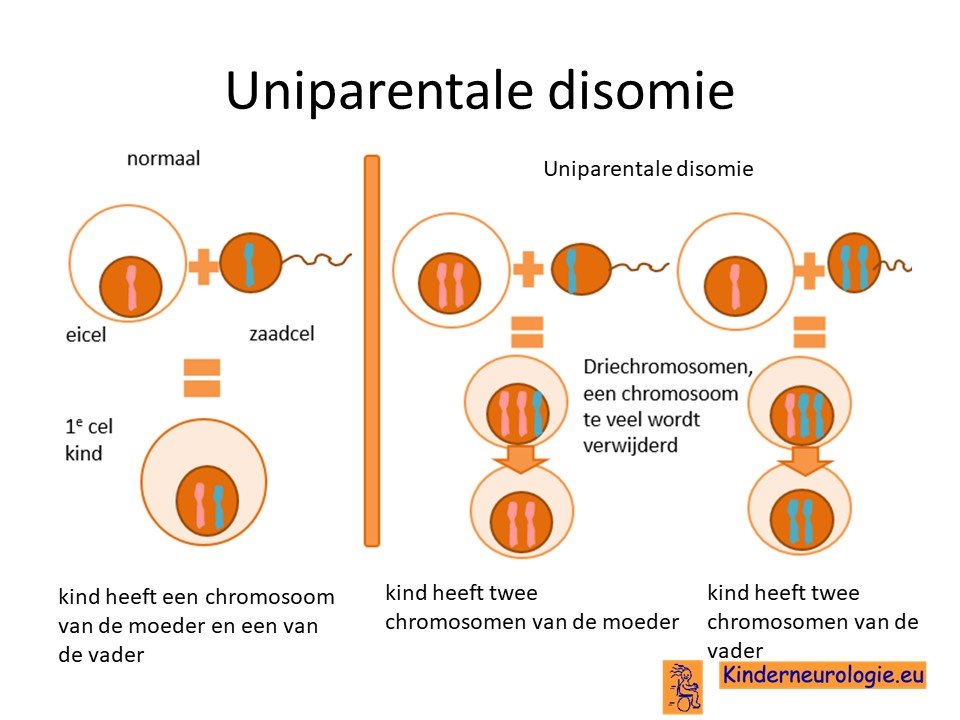
Uniparentale disomie chromosoom 15
Bij 2% van de kinderen met het Angelman syndroom is er sprake van een zogenaamde uniparentale disomie van chromosoom 15. Beide chromosomen 15 zijn dan afkomstig van de vader en niet zoals gebruikelijk één chromosoom 15 van de moeder en één chromosoom 15 van de vader. Voor een goede aanleg van de hersenen is zowel de informatie van het chromosoom van de moeder als van het chromosoom van de vader nodig. Bij kinderen met het Angelman syndroom door uniparenterale disomie mist de informatie die afgelezen wordt van het moederlijke chromosoom.
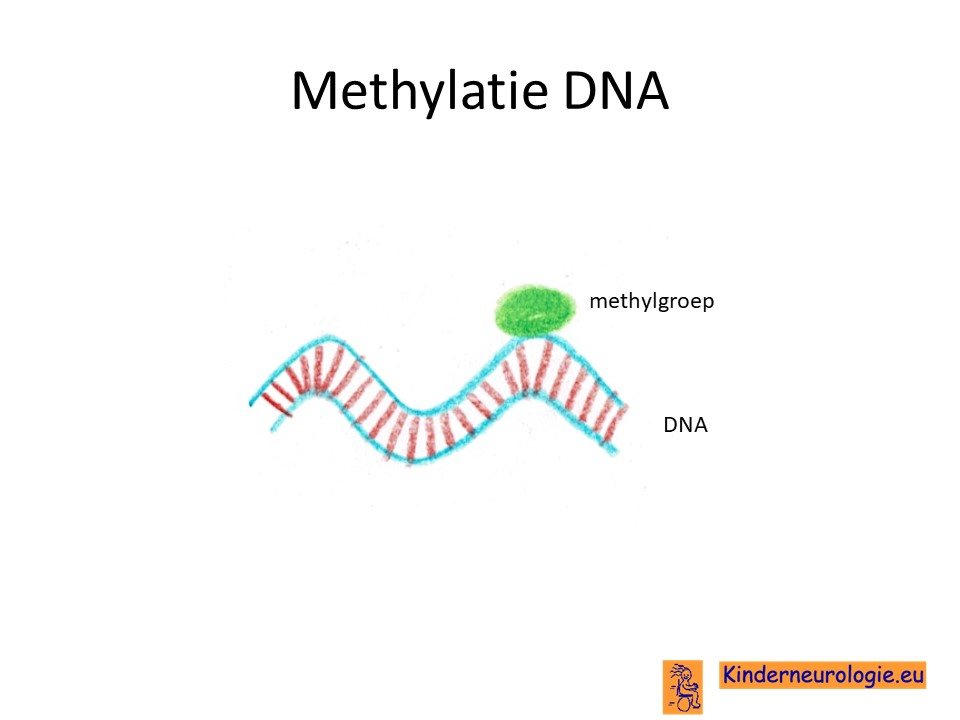
Methylatiestoornis
Om er voor te zorgen dat de informatie die op chromosoom 15 ligt goed kan worden afgelezen worden er zogenaamde hulpstukken aan het chromosoom 15 gehangen. Dit wordt methylatie genoemd. Bij een klein deel (2-3%) van de kinderen met het Angelman syndroom zit de oorzaak van het ontstaan van dit syndroom in het ontbreken van de methylatie van het chromosoom afkomstig van de moeder. Hierdoor kan de informatie niet goed afgelezen worden. Dit wordt ook wel een imprinting stoornis genoemd.
Translocatie
Bij een heel klein deel van de kinderen is chromosoom 15 die het kind van de moeder krijgt per ongeluk door midden gebroken en vervolgens op de verkeerde manier aan elkaar geplakt. De breuk in chromosoom 15 zit dan net op de plek 15q11.2 waardoor deze informatie verloren gaat en het Angelman syndroom ontstaat.
![]()
UBE3A-gen
Op het stukje chromosoom 15q11.2 ligt het UBE3A-gen. Dit gen bevat belangrijke informatie voor aanleg van de hersenen. Het bijzondere is dat dit gen zowel op het chromosoom wat afkomstig is van de moeder als afkomstig is van de vader ligt. Toch blijken kinderen alleen de informatie van het UBE3A-gen wat afkomstig is van de moeder te gebruiken voor de aanmaak van het UBE3A-eiwit. Bij kinderen met het Angelman syndroom mist deze informatie. De informatie op het vaderlijke chromosoom is wel aanwezig, maar dit kunnen de lichaamscellen blijkbaar niet gebruiken voor de aanmaak van UBE3A-eiwit.
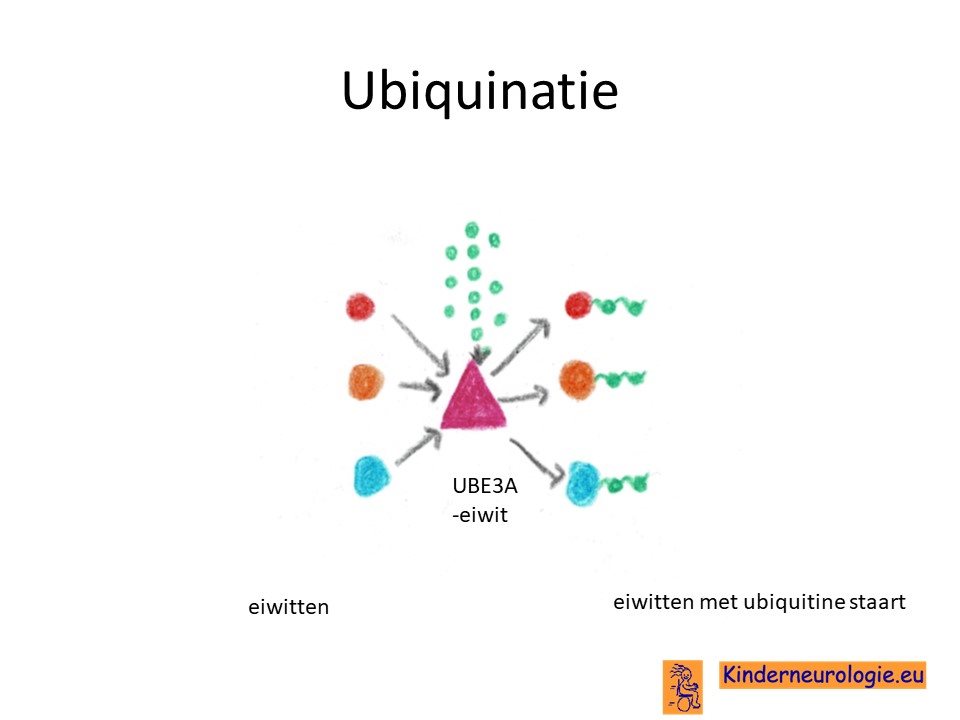
UBE3A-eiwit
Het UBE3A-eiwit speelt een belangrijke rol bij de aanleg van de hersenen. Het komt vooral voor in de kleine hersenen en in de hippocampus. Het UBE3A-eiwit hangt een zogenaamde ubiquitine groep aan allerlei verschillende soorten eiwitten. Door de aanwezigheid van dit ubiquitine eiwit verandert de werking van het eiwit. Het eiwit kan actief worden of juist inactief. Ook kan het aanhangen van het ubiquitine eiwit er voor zorgen dat het lichaam dit eiwit gaat opruimen.
Verschillende eiwitten krijgen van het UBE2A-eiwit zo'n ubiquitine groep. Een deel van deze eiwitten is bekend, het gaat om het zogenaamde Arc-eiwit, het RhoGEF-eiwit, het ephexine-5 eiwit en het sacsine-eiwit. Waarschijnlijk krijgen nog meer eiwitten van het UBE3A-eiwit een ubiquitine groep, maar dat is op dit moment nog niet bekend.
Dit proces verloopt niet goed bij kinderen met het Angelman syndroom. Hierdoor is de werking van verschillende eiwitten niet optimaal waardoor de problemen horend bij het Angelman syndroom ontstaan.
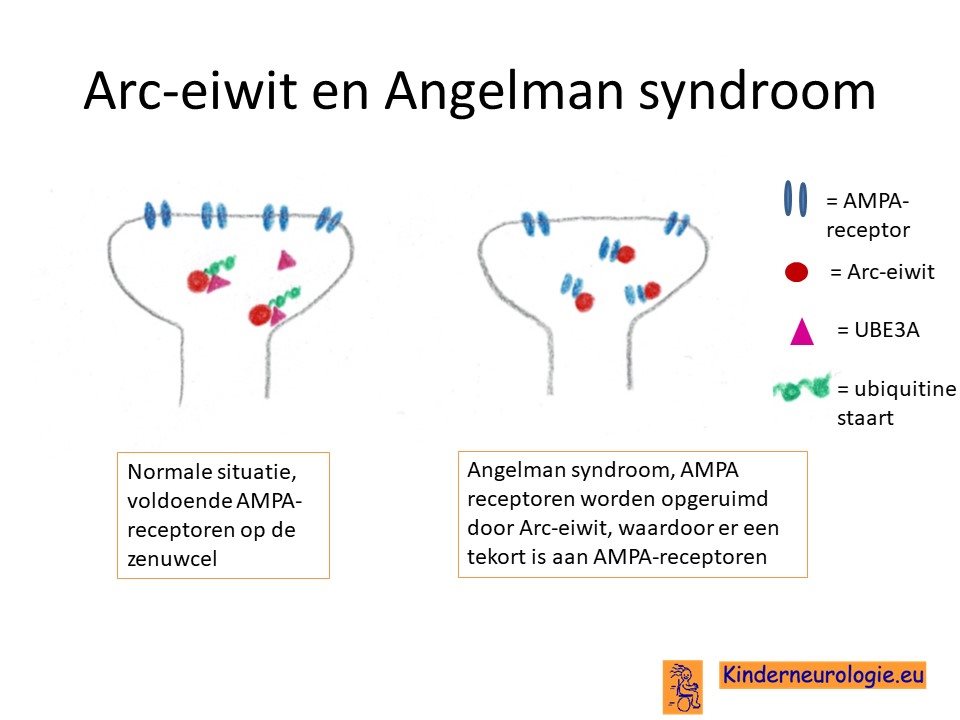
Arc-eiwit
Een van de eiwitten die niet goed werkt bij kinderen met het Angelman syndroom is het Arc-eiwit. Dit Arc-eiwit zorgt voor het opruimen van een ontvangststation voor boodschapperstofjes op zenuwcellen. Dit ontvangststation wordt de AMPA-receptor genoemd. Dit eiwit werkt te hard bij kinderen het Angelman syndroom, waardoor de AMPA-receptoren te veel worden opgeruimd en er te weinig AMPA-receptoren overblijven op de zenuwcellen. Hierdoor werken de zenuwcellen minder optimaal.
OCA2-gen
Naast het UBE3A-gen ligt het OCA2-gen. Dit gen bevat belangrijke informatie voor de aanmaak van pigment. Dit is een van de redenen dat kinderen met het Angelman syndroom als gevolg van een deletie vaak weinig pigment in hun huid, haren en iris hebben zitten.
Daarnaast komt dit ook doordat het UBE3A-gen zorgt voor het activeren van de melanocortine 1 receptor wat ook belangrijk is voor de aanmaak van pigment in de huid. Ook kinderen zonder een deletie hebben dus een grotere kans een wat lichtere huidskleur en haarkleur te hebben dan familieleden.

Wat zijn de symptomen van het Angelman syndroom?
Variatie
Er bestaat een grote variatie in de hoeveelheid en de ernst van de symptomen die verschillende kinderen met het Angelman syndroom hebben.
Dit valt van te voren niet goed te voorspellen van welke symptomen een kind last zal krijgen. Geen kind zal alle onderstaande symptomen tegelijkertijd hebben. Kinderen met een uniparenterale disomie hebben vaak minder klachten dan kinderen met een deletie.
Jouw kind is uniek
Bedenk dat onderstaande symptomen kunnen voorkomen bij jouw kind, maar ook niet allemaal zullen voorkomen. Jouw kind is uniek en veel meer dan een kind met deze aandoening. Het lezen van mogelijke symptomen die kunnen voorkomen, kan ouders het gevoel geven dat er alleen maar aandacht is voor de beperkingen van het kind. Dat is zeer zeker niet de bedoeling. Jouw kind is bijvoorbeeld lief, grappig, gevoelig, gezellig,sociaal, vindingrijk, nieuwsgierig, ondeugend, enthousiast,een zonnestraaltje, creatief en/of innemend en dat vind je niet terug in onderstaande symptomen die kunnen horen bij dit syndroom. Dat kan ook niet, want die eigenschappen maken jouw kind nu eenmaal uniek. Blijf daar vooral naar kijken en zie deze symptomen meer als achtergrondinformatie die je kunnen helpen om te begrijpen wat er met je kind aan de hand zou kunnen zijn wanneer jouw kind zich anders ontwikkelt of ergens last van heeft. Deze informatie kan jullie als ouders en hulpverleners een handvat geven wat hiervoor een mogelijke verklaring kan zijn.

Zwangerschap en bevalling
Meestal zijn er tijdens de zwangerschap en de bevalling geen bijzonderheden geweest die gemaakt hebben dat er aanwijzingen waren dat er sprake is van het Angelman syndroom. Kinderen worden geboren na een normale zwangerschapsduur, hebben een normaal geboortegewicht en een normale grootte van het hoofdje.
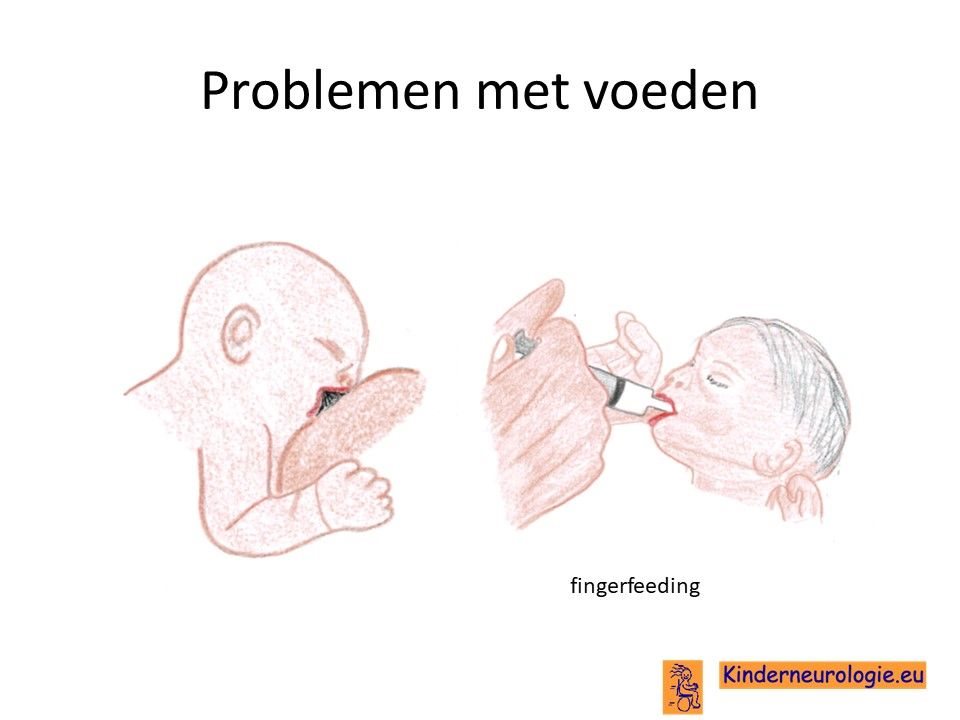
Problemen met drinken
Een groot deel van de baby’s met het Angelman syndroom heeft problemen met drinken. Ze drinken langzaam en laten de borst of speen vaak los. Kinderen vallen tijdens het voeden vaak weer in slaap. Het kost vaak veel tijd om baby’s met dit syndroom de borst of de fles te geven. Zelden is het nodig om kinderen tijdelijk sondevoeding te geven omdat zij anders niet genoeg voeding binnen krijgen en te weinig aankomen in gewicht. Baby’s met het Angelman syndroom geven gemakkelijk kleine of grotere hoeveelheden voeding terug.
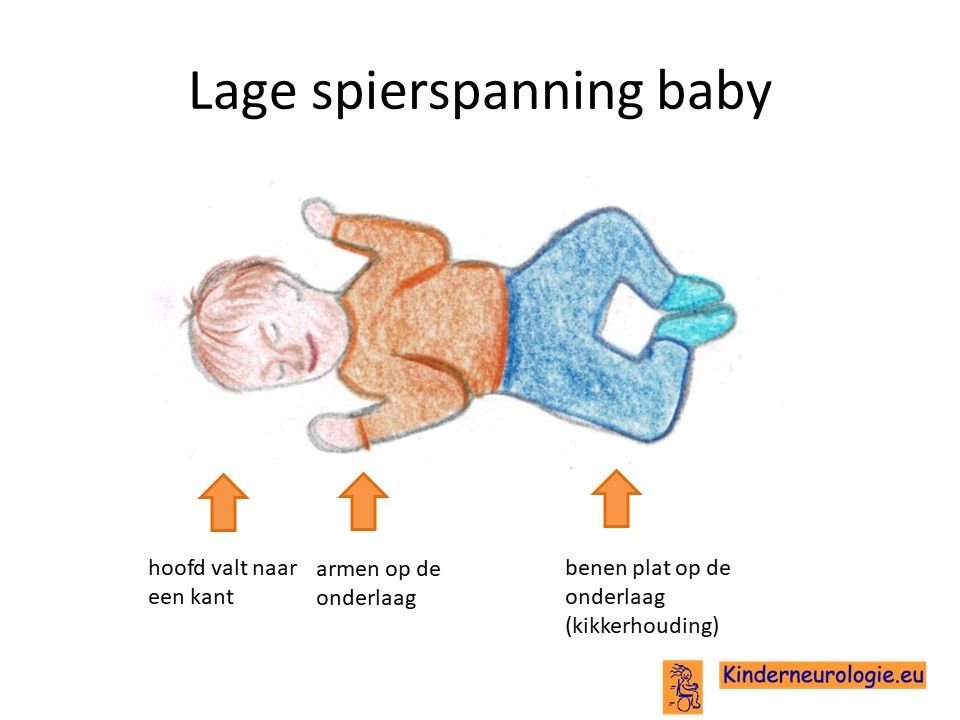
Lage spierspanning
Jonge kinderen met het Angelman syndroom voelen vaak slapper aan. Ze moeten goed vastgehouden en ondersteund worden wanneer ze opgetild worden. Door de lagere spierspanning is het voor kinderen lastig om hun hoofd op te tillen, te gaan zitten en te gaan staan. De meeste kinderen leren deze vaardigheden daarom pas op latere leeftijd dan kinderen zonder een Angelman syndroom. Bij veel kinderen met dit syndroom kunnen de gewrichten gemakkelijk overstrekt worden. Platvoeten en naar buiten gezwikte enkels komen regelmatig voor.
Onrustige baby’s
Baby’s met het Angelman syndroom zijn vaak onrustige baby’s. Zij bewegen en maaien veel met de armen en benen en hebben moeite om rust te vinden in hun lijfje. Door de onrustige bewegingen van de armen en benen, hebben kinderen moeite om in slaap te kunnen vallen.
Baby’s met het Angelman syndroom huilen weinig.
Ontwikkelingsachterstand
Kinderen met het Angelman syndroom ontwikkelen zich langzamer dan andere kinderen. Zij gaan later rollen, zitten en staan dan leeftijdsgenoten. Het merendeel van de kinderen leert zelfstandig te lopen, meestal tussen de leeftijd van 2,5 en 6 jaar oud. Kinderen met het Angelman hebben een typische manier van lopen, houterig met gebogen knieën en met de armen gebogen in de lucht om hun evenwicht te bewaren. Voor een op de tien kinderen met het Angelman syndroom is het te moeilijk om te leren zelfstandig zonder steun te lopen. Kinderen met het Angelman syndroom die epilepsie hebben, hebben vaker een grotere ontwikkelingsachterstand, dan kinderen die geen epilepsue hebben.
Spraaktaalontwikkeling
Het is voor kinderen met het Angelman syndroom heel moeilijk om te leren praten. De eerste woordjes komen vaak veel later dan bij leeftijdsgenoten. De meeste kinderen zijn in staat om klanken en enkele losse woorden te maken. Praten in zinnen is meestal niet haalbaar voor kinderen met het Angelman syndroom.
Het begrijpen van wat anderen zeggen gaat kinderen met het Angelman syndroom beter af dan het zelf praten.
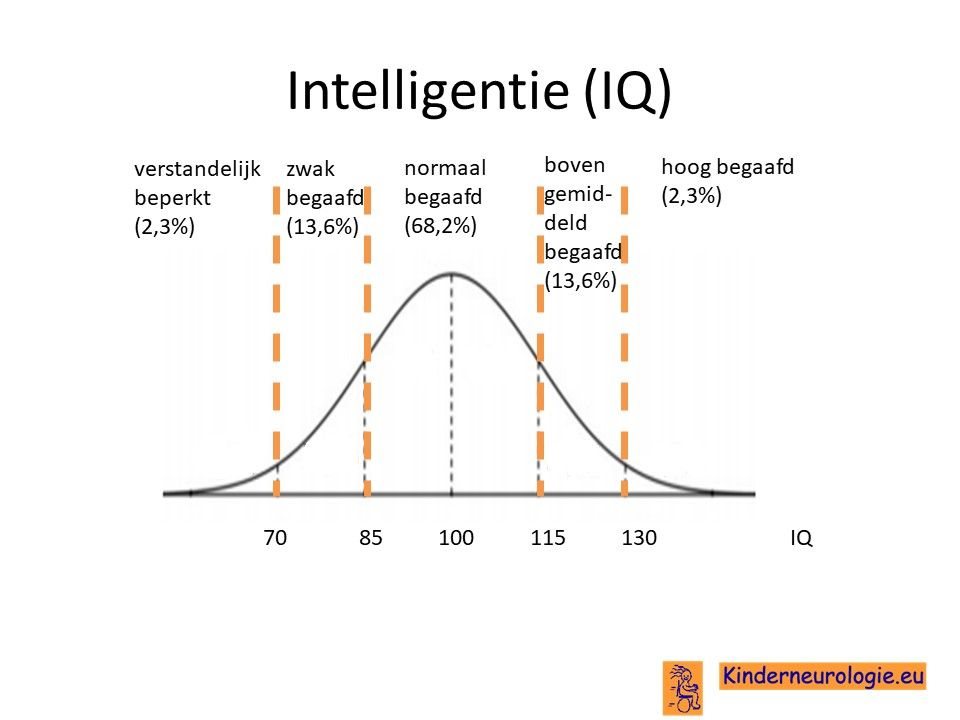
Problemen met leren
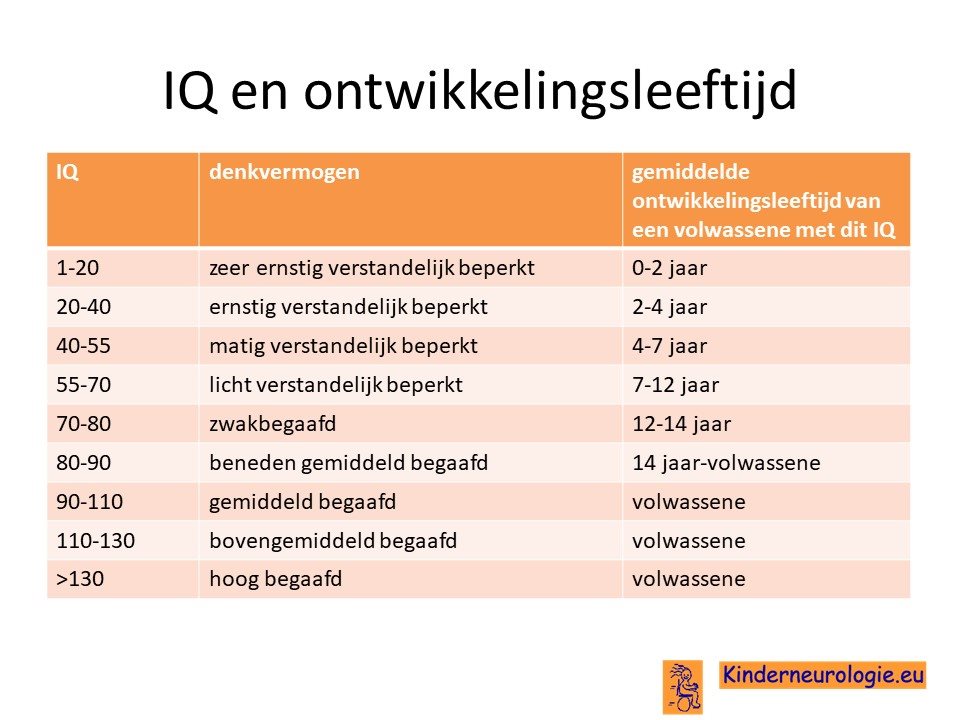
Kinderen met het Angelman syndroom hebben bijna allemaal problemen met leren. De mate van problemen met leren verschilt, sommige kinderen zijn moeilijk lerend of zeer moeilijk lerend. Kinderen met het Angelman syndroom als gevolg van een uniparenterale disomie hebben vaker een hoger IQ dan kinderen met een deletie als oorzaak. Bij een deel van de kinderen is het IQ lager dan 70, de grens waaronder wordt gesproken van een verstandelijke beperking. Een ander deel van de kinderen heeft een IQ tussen de 70 en 85, bij hen wordt er gesproken van zwakbegaafdheid.
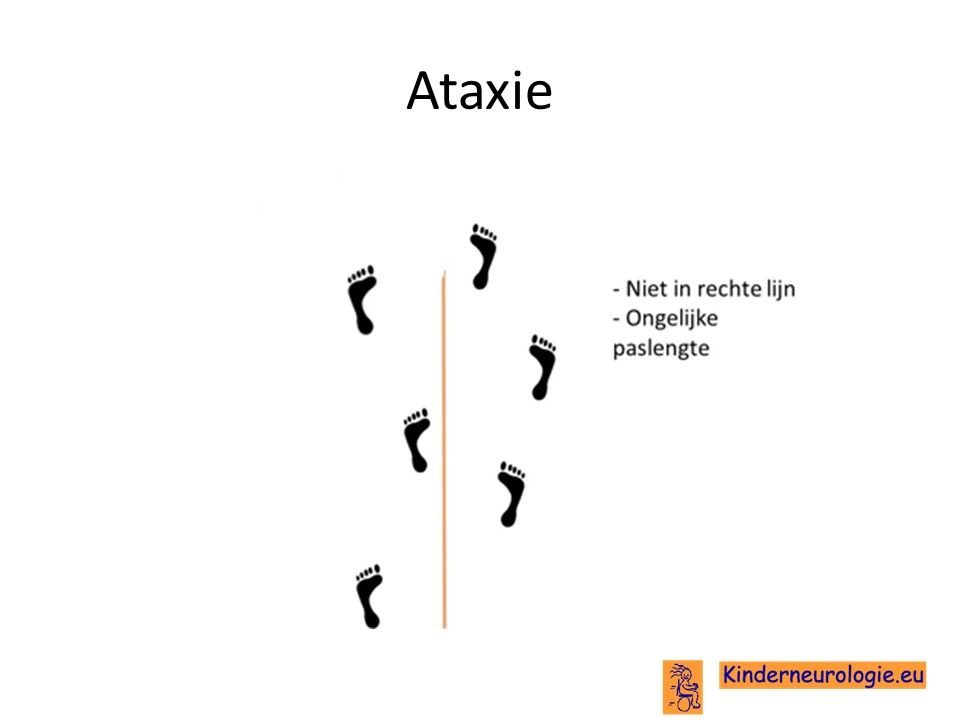
Problemen met het evenwicht
Het is voor kinderen met Angelman-syndroom vaak moeilijk om hun evenwicht te bewaren. Kinderen met het Angelman syndroom vallen gemakkelijker dan andere kinderen. Vaak zetten kinderen hun voeten wat verder uit elkaar om zo meer steun te hebben en minder snel om te vallen. Dit wordt een breedbasisch looppatroon genoemd. Kinderen houden hun armen met hun ellebogen en polsen gebogen in de lucht om hun evenwicht te kunnen bewaren. De problemen met bewaren van het evenwicht worden ataxie genoemd.
De handen kunnen een trillende beweging maken wanneer kinderen wat willen pakken. Dit trillen wordt tremor genoemd.
Hoge spierspanning
Met het ouder worden kan de spierspanning vooral in de benen geleidelijk aan hoger worden. Meestal helpt dit kinderen om te kunnen gaan staan en lopen. Een te hoge spierspanning kan zorgen voor stijfheid aan de benen, waardoor het lopen juiste weer moeilijker wordt. Een te hoge spierspanning kan maken dat kinderen de neiging krijgen om op hun tenen te gaan lopen. Dit maakt het lopen nog moeilijker. Een te hoge spierspanning wordt spasticiteit genoemd. De typische loop met gebogen knieen en heupen wordt crouch gait genoemd.

Epilepsie
Een groot deel (+/- 80%) van de kinderen met het Angelman syndroom krijgt last van epilepsieaanvallen. Meestal ontstaan deze aanvallen tussen de leeftijd van 1 en 3 jaar, bij een op de vier kinderen zelfs al voor de leeftijd van 1 jaar. Verschillende soorten aanvallen kunnen voorkomen: aanvallen met verstijven (tonische aanvallen), aanvallen met schokken van de armen en benen (clonische aanvallen), aanvallen met spierslapte (atone aanvallen) of aanvallen met staren en geen contact maken (atypische absences) en aanvallen met kleine schokjes op wisselende plaatsen (myoclonieën.). De epilepsie is vaak moeilijk onder controle te krijgen met behulp van behandeling, veel kinderen hebben dagelijks een of meerdere epilepsieaanvallen.
Kinderen met het Angelman syndroom zijn gevoelig voor het krijgen van langdurige epilepsie aanvallen. Deze aanvallen worden status epilepticus genoemd. Soms is dit duidelijk zichtbaar aan een kind, maar het kan ook weinig opvallend zijn en daardoor lastig herkenbaar. Kinderen zijn dan bijvoorbeeld wat afweziger dan anders en hebben af en toe kortdurende weinig opvallende schokjes. In dat laatste geval wordt gesproken van een non-convulsieve status epilepticus . Dit komt bij een op de vijf kinderen met Angelman syndroom voor en vaker bij kinderen met een deletie als oorzaak van het Angelman syndroom.
Ook komen koortsstuipen vaker voor bij kinderen met dit syndroom.
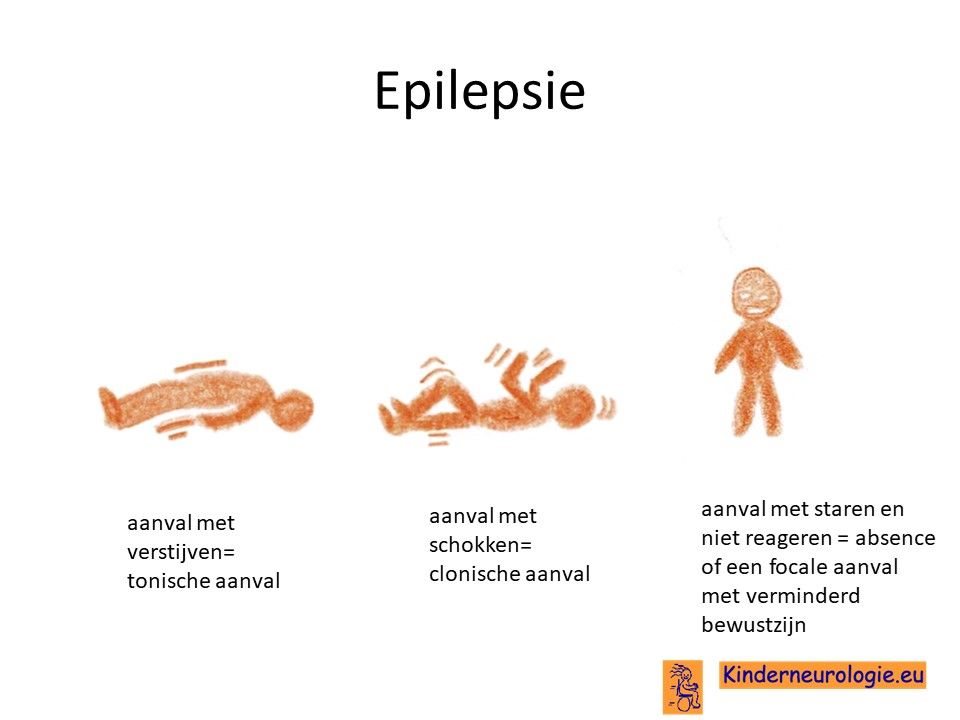
Slaapproblemen
Kinderen met het Angelman syndroom hebben vaak problemen met slapen (7-8 van de 10 kinderen). Sommige kinderen hebben moeite met het in slaap vallen en liggen langere tijd wakker voordat zij in slaap vallen. Veel kinderen zijn lichte slapers, zij worden gemakkelijk en regelmatig wakker gedurende de nacht. Ook zijn kinderen vaak vroeg in de ochtend wakker. Deze slaapproblemen hebben een invloed op het hele gezin. Kinderen met het Angelman syndroom lijken minder slaap nodig te hebben dan andere kinderen.
Deze slaapproblemen hangen voor een deel samen met verminderde melatonine aanmaak gedurende de nacht. .
Slaapproblemen kunnen het gevolg zijn van epileptische activiteit gedurende de nacht.

Lachen
Kinderen met het Angelman syndroom lachen vaak veel. Zij kunnen lachbuien hebben waarin kinderen schaterlachen zonder dat voor andere duidelijk is waarom kinderen moeten lachen. Sommige kinderen glimlachen alleen. Een deel van de kinderen heeft ook tijdens de nacht lachaanvallen.
Stereotypieën
Veel kinderen met dit syndroom maken graag bewegingen met hun armen en hun handen die vaak terug keren. Zulke bewegingen worden stereotypieën genoemd. Sommige kinderen gaan wapperen met hun handen, anderen maken draaiende bewegingen of wrijvende bewegingen over de borst heen. Deze bewegingen komen vaak voor wanneer kinderen iets heel leuks of iets spannends gaan doen. Kinderen hebben hier zelf geen last van.

Fascinatie voor water en knisperende voorwerpen
Kinderen met het Angelman syndroom zijn vaak weg van stromend water. Zij kunnen daar langere tijd mee spelen. Ook houden kinderen vaak van knisperende voorwerpen (knisperdoekjes) en glinsterende voorwerpen (spiegels).

Gedrag
Kinderen met het Angelman syndroom zijn vaak vriendelijke kinderen die veel houden van contact met andere mensen. De meeste kinderen zijn niet graag alleen, maar het liefst in de nabijheid van andere mensen. Vaak zijn kinderen nieuwsgierig en willen zij alles ontdekken. Kinderen zien vaak geen gevaar.
Tijdens de puberteit laat een deel van de kinderen agressief gedrag zien. Een deel van de kinderen gaat zich zelf verwonden door bijvoorbeeld aan haren of nagels te trekken. Dit wordt zelfverwondend gedrag genoemd. Vaak is dit een uiting van ongemak of frustratie. Ook kunnen kinderen tijdens de puberteit last krijgen van angsten.
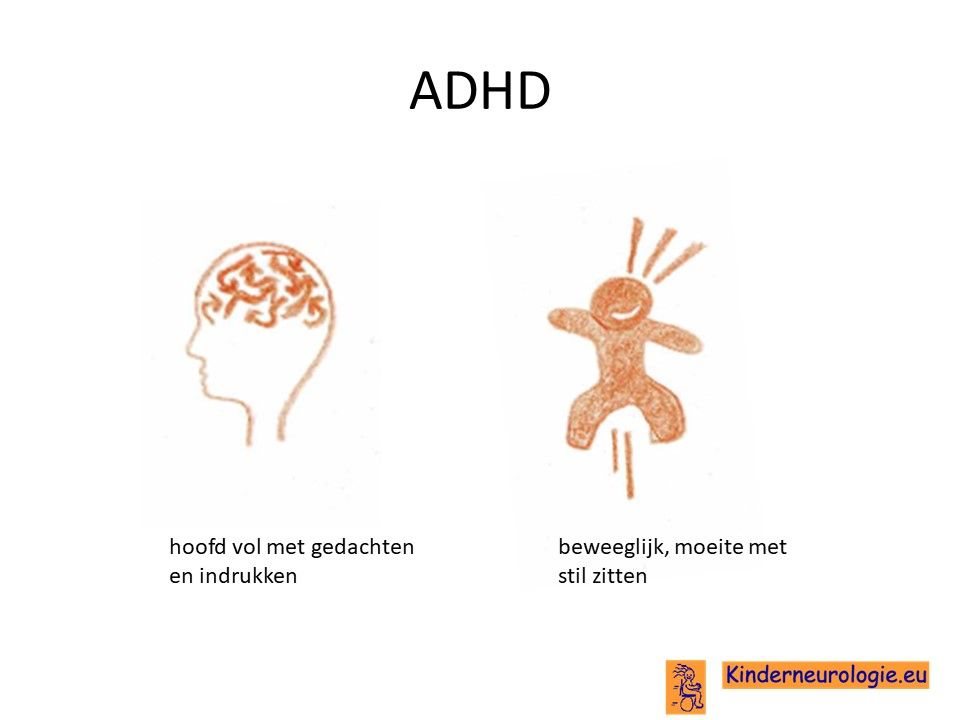
ADHD
Bij een groot deel van de kinderen met het Angelman syndroom is er sprake van ADHD. Kinderen kunnen maar korte tijd hun aandacht bij bepaald speelgoed of bij een bepaalde taak houden. Kinderen zijn gemakkelijk afgeleid door een geluid of een beweging in de omgeving. Veel kinderen met het Angelman syndroom zijn hyperactief. Zij bewegen de hele dag door en zitten zelden rustig. Vaak hebben kinderen de neiging om allerlei voorwerpen in hun mond te stoppen.
Uiterlijke kenmerken
Bij veel syndromen hebben kinderen vaak wat veranderde uiterlijke kenmerken. Hier hebben kinderen zelf geen last van, maar het kan de dokters helpen om te herkennen dat er sprake is van een syndroom en mogelijk ook van welk syndroom. Ook maakt dit vaak dat kinderen met hetzelfde syndroom vaak meer op elkaar lijken dan op hun eigen broertjes en zusjes, terwijl de kinderen toch niet familie van elkaar zijn.
Kinderen met het Angelman syndroom hebben vaak een afgeplat achterhoofdje met een diepe groef onder aan de achter kant van het hoofd. De neusrug is vaak lang en smal. De mond is vaak breed en staat open waardoor de tong naar buiten kan komen. De tanden staan vaak verder uit elkaar dan gebruikelijk. De onderkaak staat vaak meer naar voren toe dan de bovenkaak.

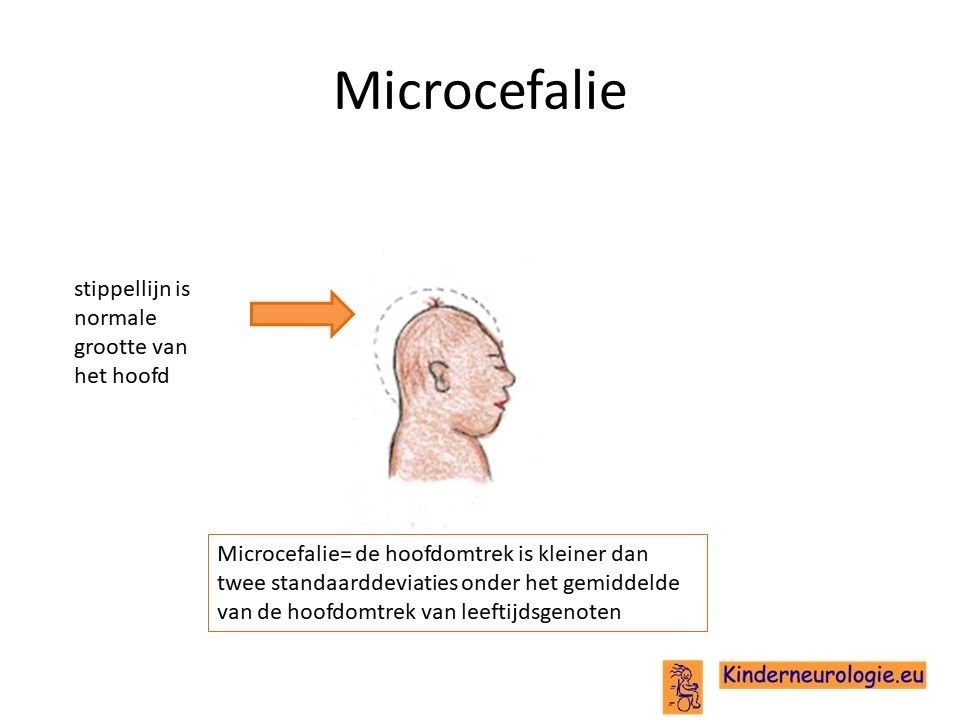
Klein hoofd
Bij de geboorte hebben kinderen met het Angelman syndroom een normale grootte van hun hoofd. Na de geboorte groeit het hoofd van kinderen met het Angelman syndroomniet zo hard als bij leeftijdsgenoten. Geleidelijk aan ontstaat hierdoor een klein hoofd. Een te klein hoofdje wordt een microcefalie genoemd. Omdat dit kleine hoofd pas met het ouder worden ontstaat en niet al bij de geboorte aanwezig is, wordt gesproken van een secundaire microcefalie. Bij vier op de vijf kinderen met het Angelman syndroom is er sprake van een microcefalie. Microcefalie komt vaker voor bij kinderen met een grote deletie dan bij kinderen met een uniparenterale disomie.
Lengte
De lengte van kinderen met het Angelman syndroom is normaal.
Lichte huid en haren
Kinderen die het Angelman syndroom hebben als gevolg van een deletie hebben vaak een lichte huid met blonde haren en blauwe ogen. Door deze lichte huid hebben kinderen met het Angelman sneller last van verbranding door zonlicht.
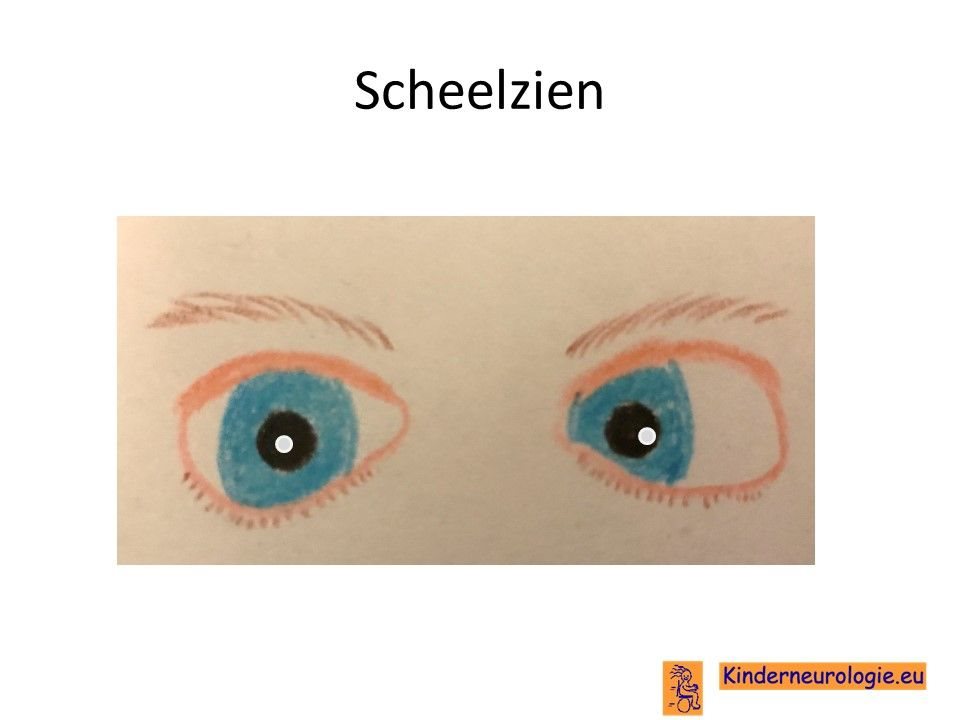
Problemen met zien
Scheelzien komt vaak voor bij kinderen met het Angelman syndroom. Vaak staan een of beide ogen meer naar buiten in de richting van de oren toe. Bij kinderen met een deletie hebben de ogen hebben vaak een lichte kleur. Een groot deel van de kinderen is slechtziend. Zowel bijziendheid als verziendheid komen vaker voor. Een deel van de kinderen met het Angelman syndroom heeft de neiging om veel in de ogen te wrijven. Hierdoor kan beschadiging van het hoornvlies ontstaan.
Problemen met horen
Het hebben van oorontstekingen kan zorgen voor vocht achter het trommelvlies. Hierdoor kunnen kinderen slechter horen.

Kwijlen
Kinderen met het Angelman syndroom hebben gemakkelijk last van kwijlen. Deels komt dit doordat zij hun mond vaak open hebben staan. Het speeksel kan dan gemakkelijk via de mond naar buiten toe lopen.
Alles in de mond stoppen
Kinderen met het Angelman syndroom mogen graag iets in hun mond stoppen, zoals hun vingers, handen of andere voorwerpen. Deze voorwerpen in de mond zorgen vaak voor toename van kwijlen. Het stoppen van voorwerpen in de mond wordt ook wel mouthing behaviour genoemd.

Bijzonder eetgedrag
Oudere kinderen met het Angelman syndroom hebben vaak bijzonder eetgedrag. Ze stoppen alles in de mond en proberen dit op te eten. Dit kunnen zowel eetbare producten zijn als niet eetbare producten. Kinderen kunnen ook kauwbewegingen maken waardoor het lijkt alsof zij iets aan het eten zijn zonder dat zij iets in de mond hebben. Kinderen met het Angelman syndroom voelen niet goed aan dat zij genoeg gegeten hebben. Zij kunnen dus gemakkelijk overeten.
Overgewicht
Vanaf de puberteit hebben kinderen met het Angelman syndroom door de dit afwijkende eetgedrag de neiging om gemakkelijk dikker te worden waardoor overgewicht kan ontstaan.
Tandenknarsen
Een deel van de kinderen met het Angelman syndroom doet aan tandenknarsen. Zij bewegen hun tanden en kiezen over elkaar heen waardoor een typisch knarsend geluid ontstaat.
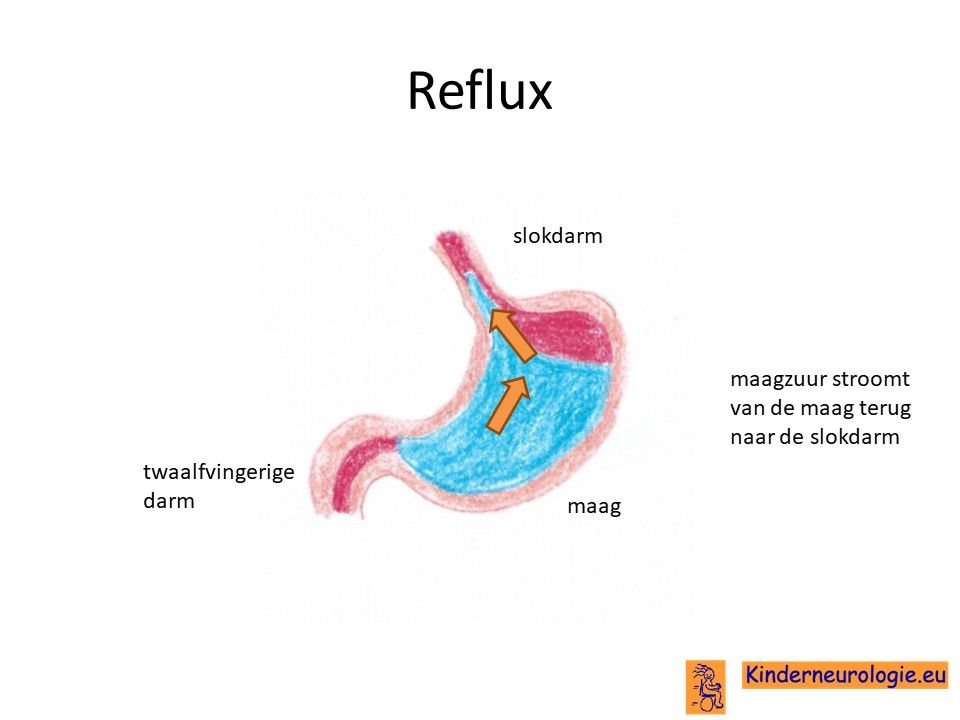
Reflux
Een deel van de kinderen met het Angelman syndroom heeft last van reflux. Maagzuur en/of voeding stromen vanuit de maag terug naar de slokdarm of de mond. Dit kan zorgen voor een vieze smaak en pijnklachten waardoor kinderen onrustig worden. Ook kan reflux zorgen dat kinderen moeten spugen of minder zin hebben in eten. Reflux kan zorgen voor een vieze geur uit de mond. Ook kan reflux zorgen voor het ontstaan van luchtweginfecties en aantasting van het tandglazuur.
Verstopping van de darmen
Verstopping van de darmen komt vaak voor bij kinderen met het Angelman syndroom. De ontlasting komt dan niet elke dag en is vaak hard waardoor kinderen moeite hebben met poepen. Dit kan buikpijnklachten geven en zorgen voor een bolle buik. Ook kan de eetlust hierdoor minder worden.
Zindelijkheid
Het duurt vaak langer dan gebruikelijk voordat kinderen met dit syndroom zindelijk worden. De meeste kinderen worden dit uiteindelijk wel uit zichzelf.

Puberteit
Kinderen met het Angelman syndroom maken net als kinderen zonder dit syndroom een normale puberteit door.
Niet goed tegen warmte kunnen
Kinderen met het Angelman syndroom kunnen vaak niet goed tegen een warme omgevingstemperatuur.
Vatbaarder voor infecties
Kinderen met het Angelman-syndroom zijn op jonge leeftijd vatbaarder voor het krijgen van infecties. Regelmatig komen luchtweginfecties of oorontstekingen voor.
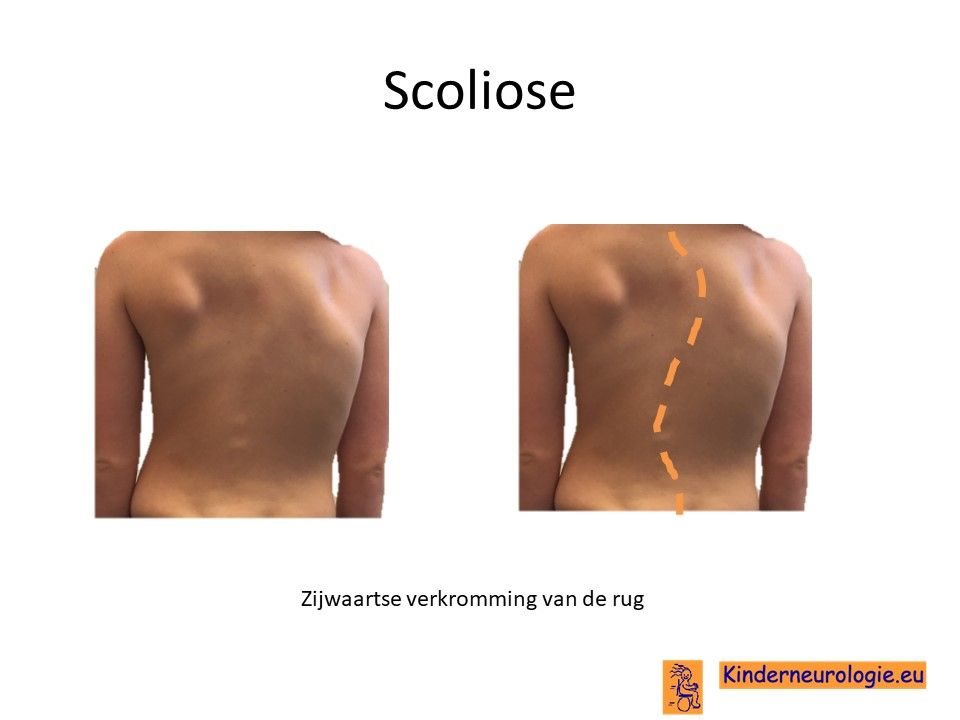
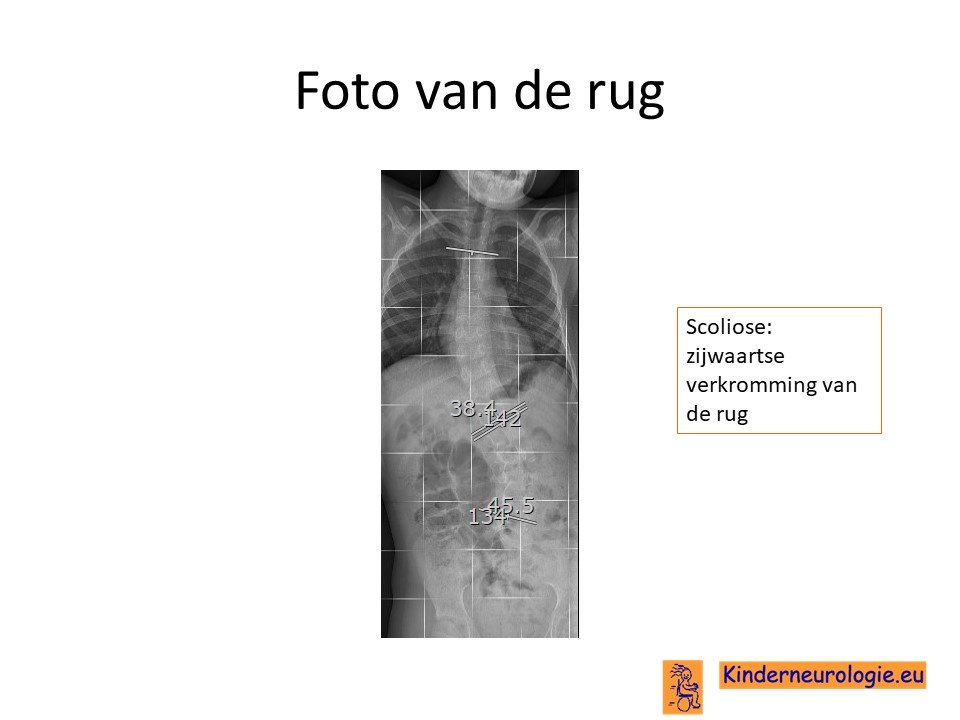
Scoliose
Een groot deel van de kinderen met het Angelman syndroom krijgt een zijwaartse verkromming van de rug. Dit wordt een scoliose genoemd. Deze scoliose is vaak in lichte mate al op jonge leeftijd aanwezig en neemt toe tijdens de puberteit. Een ernstige scoliose kan zorgen voor pijnklachten in de rug en problemen met zitten en staan. Ook kunnen er problemen ontstaan met de ademhaling als gevolg van de scoliose.
Hoe wordt de diagnose Angelman syndroom gesteld?
Verhaal en onderzoek
Op grond van het verhaal van een kind met een ontwikkelingsachterstand, een lage spierspanning, typische uiterlijke kenmerken en achter blijven van de groei van het hoofdje kan worden vermoed dat er sprake is van het Angelman syndroom. Er zijn echter ook andere syndromen die veel op het Angelman syndroom kunnen lijken zoals het Rett-syndroom, het Mowat-Wilson syndroom, ATRX-syndroom, het Christian syndroom of het Pitt-Hopkins syndroom. Aanvullende diagnostiek zal nodig zijn om de diagnose Angelman syndroom te stellen.
Bloedonderzoek
Bij standaard bloedonderzoek worden geen bijzonderheden gezien bij kinderen met het Angelman syndroom.
Genetisch onderzoek
Wanneer aan de diagnose gedacht wordt, kan door middel van gericht genetisch onderzoek op bloed naar het voorkomen van het Angelman syndroom. Hiervoor moet een speciale methylatietest worden aangevraagd. Deze test is bij 8 van de 10 kinderen met het Angelman syndroom afwijkend.
Wanneer de methylatietest niet afwijkend is, kan gericht worden gezocht naar foutjes op het UBE3A-gen.
Wanneer de methylatietest afwijkend is zal het laboratorium met behulp van een zogenaamde MLPA test kunnen achterhalen welke fout in het erfelijk materiaal de oorzaak is van het Angelman syndroom (deletie, uniparenterale disomie, methylatiedefect of translocatie).
Vaak worden ook alle chromosomen tegelijkertijd onderzocht (zogenaamd Array onderzoek). Wanneer er sprake is van een deletie kan soms op deze manier de diagnose Angelman syndroom worden gesteld, de andere vormen kunnen worden gemist.
Tegenwoordig kan door middel van een nieuwe genetische techniek (exome sequencing genoemd) mogelijk ook deze diagnose gesteld kunnen worden zonder dat er specifiek aan gedacht was of naar gezocht is. Deze test kan echter niet alle vormen van Angelman syndroom opsporen.
MRI-scan
Bij kinderen met een ontwikkelingsachterstand zal vaak een MRI scan gemaakt worden om te kijken of er bijzonderheden aan de hersenen te zien zijn. Bij een groot deel van de kinderen kinderen met het Angelman syndroom worden geen of nauwelijks afwijkingen gezien op de MRI scan. Soms is te zien dat het volume van de grote hersenen kleiner is dan gebruikelijk. Ook kan de aanleg van het geleidingslaagje rondom de zenuwuitlopers in de hersenen iets vertraagd verlopen. Deze afwijkingen zijn heel aspecifiek en kunnen ook bij allerlei andere soorten syndromen worden gezien.
Stofwisselingsonderzoek
Kinderen met een ontwikkelingsachterstand krijgen vaak stofwisselingsonderzoek van bloed en urine om te kijken of er sprake is van een stofwisselingsziekte die verklarend is voor de ontwikkelingsachterstand. Bij kinderen met het Angelman syndroom worden hierbij geen bijzonderheden gezien.
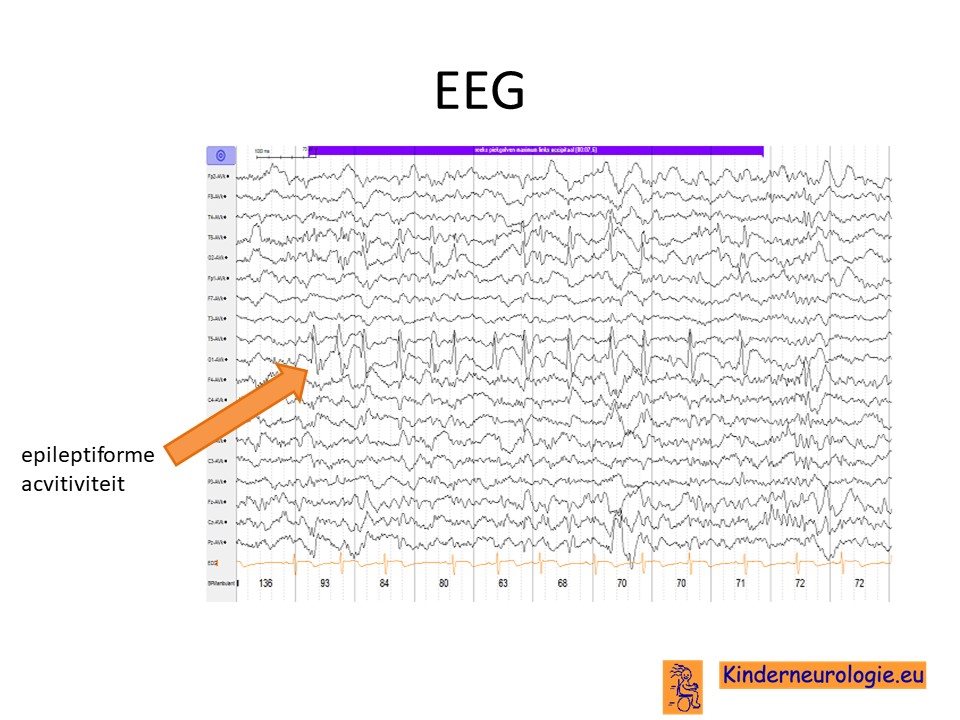
EEG
Bij vermoeden op epilepsie zal vaak een EEG gemaakt worden. Een groot deel van de kinderen met het Angelman syndroom heeft zogenaamde trage hooggevolteerde activiteit op het EEG met daarin pieken. Dit is vrij kenmerkend voor kinderen met het Angelman syndroom. Het hebben van EEG afwijkingen heeft geen duidelijke relatie met het voorkomen van epilepsieaanvallen. Er zijn kinderen met epileptiforme afwijkingen op het EEG die geen last hebben van epilepsieaanvallen.
Oogarts
Kinderen met het Angelman syndroom worden altijd een keer door een oogarts gezien om te kijken of er problemen zijn met zien.

KNO-arts
De KNO-arts kan beoordelen of er sprake is van problemen met het gehoor.
Foto van de botten
Wanneer er sprake is van een verkromming van de wervelkolom zal vaak een foto van de botten gemaakt worden om de mate van verkromming vast te leggen en om te kijken hoe de wervels van de rug zijn aangelegd.
DEXA-scan
Door middel van en dexa-scan kan de botdichtheid worden gemeten.
Hoe wordt het Angelman syndroom behandeld?
Geen genezing
Er is geen behandeling die het Angelman syndroom kan genezen. De behandeling is er op om het kind er zo goed mogelijk te leren om gaan met de symptomen die horen bij het Angelman syndroom en om problemen zoals overgewicht te voorkomen.
Kinderarts
In Nederland wordt door de zorg voor kinderen met een zeldzaam syndroom vaak gecoordineerd door de kinderarts in de eigen woonomgeving. Daarnaast kunnen kinderen ook begeleid worden door een speciale kinderarts die zich gespecialiseerd heeft in de zorg voor kinderen met een aangeboren en vaak zeldzame aandoeningen. Deze kinderarts heet kinderarts EAA: kinderarts voor erfelijke en aangeboren aandoeningen. In steeds meer ziekenhuizen in Nederland werken kinderartsen EAA. De kinderarts EAA stemt met de eigen kinderarts in de woonomgeving van het kind af hoe de zorg voor het kind zo optimaal mogelijk kan verlopen. Ook kan de kinderarts EAA gespecialiseerde kinderartsen om hulp vragen zoals een kindercardioloog bij hartproblemen, een kinderlongarts voor longproblemen, een kinderMDL-arts of kinderchirurg voor darmproblemen, een kindernefroloog of kinderuroloog voor problemen met de nieren en/of plassen, een kinderneuroloog voor kinderen met epilepsie of bewegingsstoornissen, een kinderorthopeed voor kinderen met afwijkingen van de botten, een kinderendocrinoloog voor kinderen met afwijkingen van de hormomen en/of een kinderdermatoloog voor kinderen met huidafwijkingen.

Aanvalsbehandeling epilepsie
De meeste epilepsieaanvallen gaan vanzelf over binnen enkele minuten. Omstanders hoeven dan niets te doen om de aanval te doen stoppen. Het is belangrijk om zo rustig mogelijk te blijven en het kind zo veel mogelijk met rust te laten.
Wanneer een aanval na 5 minuten nog niet vanzelf gestopt is, dan zal vaak geadviseerd worden om medicijnen te geven om een aanval te doen stoppen. De behandelende arts zal altijd aangeven welk tijdstip voor een bepaald kind het beste is. Medicijnen die gebruikt kunnen worden voor het stoppen van een aanval zijn diazepam rectiole (Stesolid®), midazolam neusspray, midazolam rectiole, lorazepam of clonazepam druppels.
Het effect van deze medicijnen ontstaat na enkele minuten. Nadien zal het kind meestal in slaap vallen, soms ook niet.

Behandeling epilepsie
Met behulp van medicijnen wordt geprobeerd om de epilepsieaanvallen zo veel mogelijk te voorkomen en het liefst er voor te zorgen dat er helemaal geen epilepsieaanvallen meer voorkomen. Het is vaak moeilijk om de epilepsie helemaal onder controle te krijgen, vaak zijn combinaties van medicijnen nodig. Meestal wordt gezocht naar een combinatie van medicijnen waarbij zo min mogelijk epilepsieaanvallen voorkomen en waarbij kinderen ook zo min mogelijk bijwerkingen hebben van de medicijnen.
Verschillende soorten medicijnen kunnen gebruikt worden om de epilepsie onder controle te krijgen. Er bestaat geen duidelijk voorkeursmedicijn. Medicijnen die vaak gebruikt worden zijn natriumvalproaat (Depakine ®), levetiracetam (Keppra ®), clobazam (Frisium ®), clonazepam (Rivotril®), lamotrigine (Lamictal ®), ethosuximide (Ethymal ®), topiramaat (Topamax®) en zonisamide (Zonegran®). Een deel van de kinderen, maar zeker niet alle kinderen, reageert minder goed op het gebruik van carbamazepine (Tegretol®), oxcarbazepine (Trileptal®) en vigabatrine (Sabril ®).
Bij een deel van de kinderen zal het niet lukken om de epilepsieaanvallen met medicijnen onder controle te krijgen. Er bestaan ook andere behandelingen die een goed effect kunnen hebben op de epilepsie,zoals een ketogeen dieet, een nervus vagusstimulator, of een behandeling met methylprednisolon. Ook een combinatie van deze behandelingen met medicijnen die epilepsie onderdrukken is goed mogelijk.

Slaapproblemen
Een vast slaapritueel en een vast slaappatroon kunnen kinderen helpen om beter te kunnen slapen. Het lage dosering van het medicijn melatonine ( maximaal 0,3 mg) kan helpen om beter in slaap te kunnen vallen. Omdat kinderen met het Angelman syndroom melatonine traag afbreken, moeten zij niet een te hoge dosering melatonine krijgen. Dit zorgt anders voor vermoeidheid overdag. Slaapmiddelen worden liever niet gegeven aan kinderen omdat kinderen hier aangewend raken en niet meer zonder deze medicatie kunnen. Soms wordt het medicijn promethazine gebruikt om kinderen beter te kunnen laten slapen. Bij volwassenen met slaapproblemen kan het antidepressivum mirtazepine helpen om beter te kunnen slapen
Het is altijd belangrijk om uit te sluiten dat epilepsie de oorzaak is van de slaapproblemen, in geval van epilepsie is epilepsie behandeling nodig.

Fysiotherapie
Een fysiotherapeut kan ouders tips en adviezen geven hoe ze hun kindje zo goed mogelijk kunnen stimuleren om er voor te zorgen dat de ontwikkeling zo optimaal als mogelijk verloopt. Ook kan de fysiotherapeut tips geven hoe ouders hun kind kunnen stimuleren om te bewegen. Het is belangrijk dat kinderen regelmatig bewegen om zo de spierkracht van kinderen te verbeteren. Zwemmen is heel goed voor kinderen met het Angelman syndroom en de meeste kinderen beleven hier veel plezier aan.

Logopedie
Een logopediste kan tips en adviezen geven indien er problemen zijn met zuigen, drinken, kauwen of slikken. Sommige kinderen hebben baat bij een speciale speen (special need speen) waardoor het drinken uit de fles beter verloopt. Moeders kunnen borstvoeding kolven, zodat kinderen op deze manier toch borstvoeding als voeding kunnen krijgen via de fles.
Ook kan de logopediste helpen om de spraakontwikkeling zo goed mogelijk te stimuleren. Daarnaast wordt kinderen geleerd te communiceren door middel van gebaren of pictogrammen. Op die manier kunnen kinderen zich leren uitdrukken ook als ze nog geen woorden kunnen gebruiken. Dit helpt hen om frustratie te voorkomen. Er bestaan speciale methodes (PODD genoemd) die ouders en anderen kunnen helpen om kinderen te leren communiceren. Er bestaan speciale PODD-boeken met daarin plaatjes van allerlei woorden.
Oudere kinderen hebben vaak baat bij een spraakcomputer.

Voorkomen overgewicht
Kinderen met het Angelman syndroom krijgen gemakkelijk last van overgewicht. Het is daarom belangrijk om al vanaf jonge leeftijd op te letten dat kinderen niet te veel gaan eten. Pubers kennen geen rem op het eten en hebben hun ouders nodig om aan te geven dat zij voldoende hebben gegeten.
Diëtiste
Een diëtiste kan berekenen hoeveel calorieën een kind met het Angelman syndroom per dag nodig heeft. Op deze manier kan voorkomen worden dat kinderen overgewicht ontwikkelen.

Ergotherapie
Een ergotherapeut kan tips en adviezen geven hoe de verzorging en de dagelijks activiteiten van een kind zo soepel mogelijk kunnen verlopen. Ook kan de ergotherapeut advies geven over materialen die de ontwikkeling van een kind kunnen stimuleren.
Wanneer schrijven lastig wordt, kan het bijvoorbeeld helpen om te schrijven met een dikkere pen. Ook bestaat er bestek met dikkere handvaten die gemakkelijker vast te houden zijn en zijn er hulpmiddelen om kleding zelf aan te kunnen trekken als dat lastig gaat.

Revalidatiearts
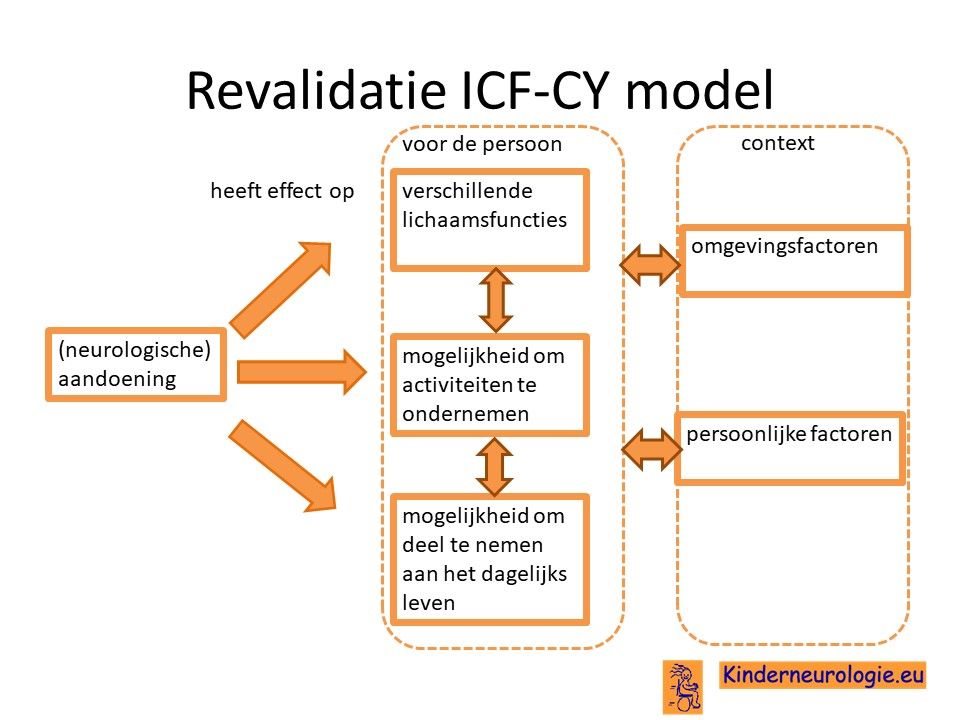
De revalidatiearts coördineert de verschillende behandelingen en vervolgt de ontwikkeling van kinderen met een ontwikkelingsachterstand. Bij problemen wordt gekeken wat voor oplossing er voor deze problemen te bedenken is. Vaak doet de revalidatiearts dit aan de hand van ICF-CY model. Er wordt gekeken wat het effect is van de aandoening op de verschillende lichaamsfuncties van het kind, de mogelijkheid om activiteit te ondernemen (bijvoorbeeld eten, aankleden, spelen) en de mogelijkheden om deel te nemen aan het dagelijks leven. De revalidatiearts denkt samen met een team mee welke oplossingen er te bedenken zijn voor een bepaald probleem.
De revalidatiearts geeft ook adviezen voor aangepaste schoenen of het gebruik van bijvoorbeeld spalken.
De revalidatiearts geeft ook adviezen voor het juiste onderwijs van het kind. Sommige kinderen gaan naar de school die verbonden is aan het revalidatiecentrum.
Ook voor jongere kinderen bestaan op het revalidatiecentrum vaak groepjes waarin de kinderen gedurende een dagdeel therapie krijgen waarin hun ontwikkeling gestimuleerd wordt.
Dagopvang
Vanaf de leeftijd van 2 maanden kunnen kinderen die niet naar een reguliere kinderdagopvang kunnen, naar een speciale kinderdagopvang toe gaan. Er bestaat speciale therapeutische peutergroepen in revalidatiecentra, of dagopvang in een orthopedagogisch dagcentrum (ODC) of in een medische kinderdagcentrum (MKD). Het hangt van de problemen die het kind ervaart af (zoals epilepsie of gedragsproblemen), welke vorm van dagopvang het meest geschikt is. Aanmelding voor een ODC of een MKD verloopt via de gemeente (vaak cia het centrum jeugd en gezin, via het jeugdteam of via het sociaal wijkteam). Aanmelding voor een therapeutische peutergroep in een revalidatiecentrum verloopt via de revalidatiearts.
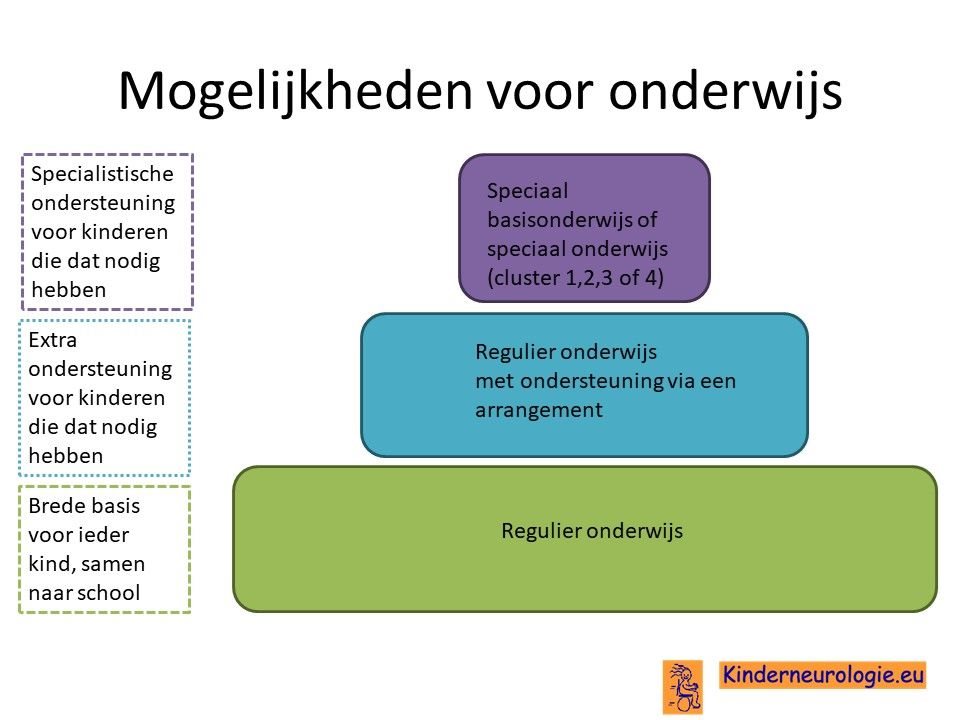
School
Een deel van de kinderen met het Angelman syndroom bezoekt regulier onderwijs met extra ondersteuning. Vaak volgen kinderen hun eigen leerlijn. Een ander deel van de kinderen bezoekt speciaal onderwijs. In het speciaal onderwijs zijn de klassen kleiner en kan het lesprogramma meer afgestemd worden op de mogelijkheden van het kind. Voor een deel van de kinderen is het niet haalbaar om onderwijs te volgen. Zij gaan naar een dagcentrum waar kinderen een dagprogramma volgen.Het LWOE kan leerkrachten adviezen geven hoe kinderen met epilepsie op school het beste begeleid kunnen worden.
Orthopedagoog
Een orthopedagoog kan ouders tips en adviezen geven hoe om gaan met problemen met bijvoorbeeld boos worden of zich zelf verwonden.
AVG-arts
Een Arts voor verstandelijke gehandicapten kan ouders helpen hoe zij hun kind zo goed mogelijk kunnen begeleiden en stimuleren. Ook kan de AVG-arts meedenken wat de oorzaak is van frustratie, agressie of zelfverwonding. Dit is niet altijd makkelijk te achterhalen omdat het voor de jongeren met Angelman heel moeilijk is om zichzelf uit te drukken.
Kinder- en jeugdpsychiater
Een kinder- en jeugdpsychiater kan advies geven hoe om te gaan met gedragsproblemen zoals ADHD, plotse woede-uitbarstingen, zelfverwonding en angst. Soms is ondersteunende medicatie nodig zoals risperidon bij prikkelovergevoeligheid, methylfenidaat voor hyperactiviteit of amitriptyline bij angst.
Oogarts
Een deel van de kinderen heeft een bril nodig om goed te kunnen zien. Wanneer kinderen scheel kijken, dan kan het nodig zijn om een oog een aantal uur per dag af te plakken, om op die manier te voorkomen dat kinderen een lui oog ontwikkelen.

KNO-arts
Bij kinderen met frequente middenoorinfecties kunnen buisjes nodig zijn om nieuwe middenoorontstekingen te voorkomen.
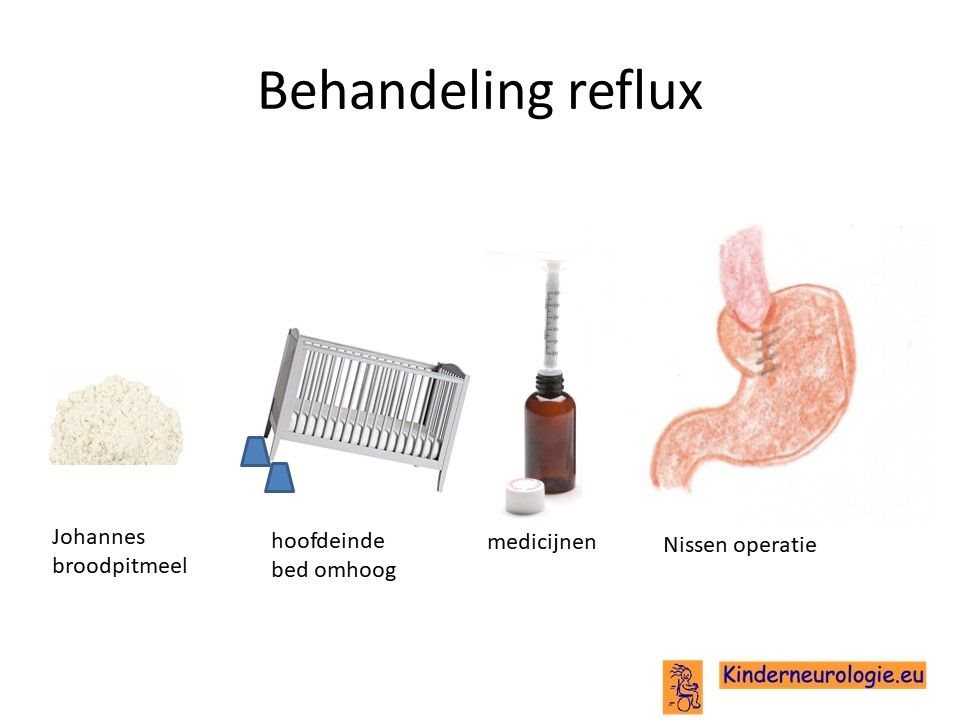
Reflux
Reflux kan er ook voor zorgen dat kinderen slecht eten. Door de voeding in te dikken met johannesbroodpitmeel kan de voeding minder gemakkelijk terug stromen van de maag naar de slokdarm. Ook zijn er medicijnen die de maaginhoud minder zuur kunnen maken waardoor de slokdarm minder geprikkeld wordt bij terugstromen van de maaginhoud. Medicijnen die hiervoor gebruikt worden zijn ranitidine en omeprazol, soms esomeprazol. Indien dit allemaal niet voldoende is, kan een operatie nodig zijn waarbij de overgang van de slokdarm naar de maag nauwer wordt gemaakt, waardoor de voeding ook minder gemakkelijk terug kan stromen.
Tandenpoetsen
Regelmatig poetsen van de tanden en het gebruik van mondwater kan helpen om gaatjes te voorkomen.
Tandarts
Het is belangrijk dat kinderen met het Angelman syndroom vanaf jonge leeftijd regelmatig gezien worden door de tandarts. Er bestaan speciale tandartsen die zich gespecialiseerd hebben in de tandheelkundige zorg van kinderen met een ontwikkelingsachterstand omdat dit vaak speciale aanpak en extra tijd vraagt.
De tandarts bekijkt of een fluor behandeling nodig is om gaatjes in de tanden en kiezen als gevolg reflux en het stoppen van voorwerpen in de mond te voorkomen.

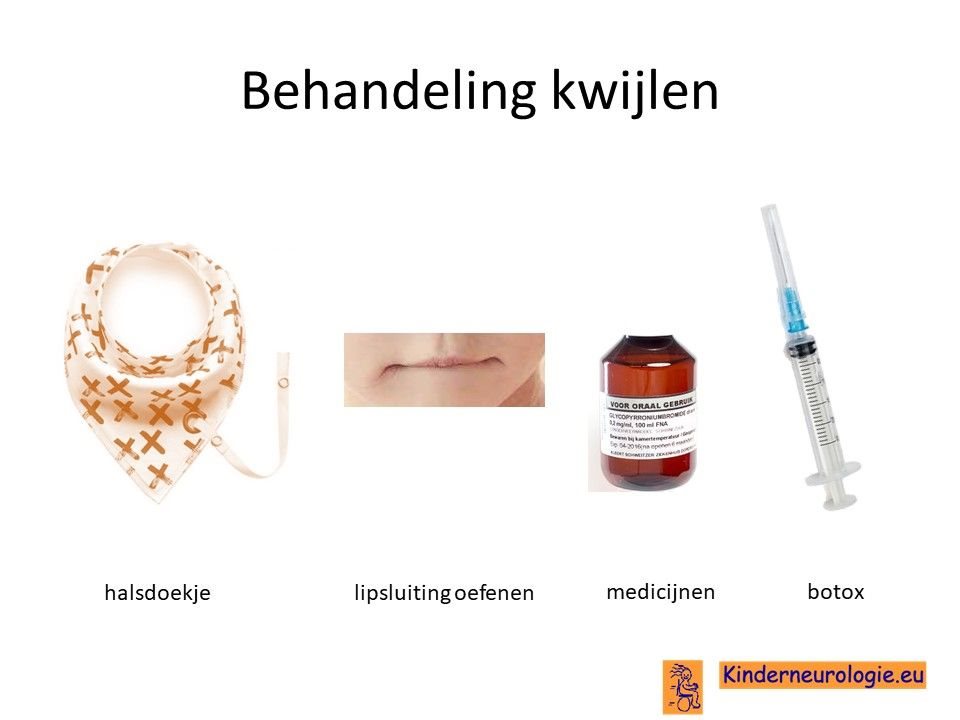
Kwijlen
Kwijlen kan verminderen door kinderen er bewust van te maken dat ze hun speeksel moeten doorslikken. Ook kunnen oefeningen waarbij geoefend wordt om de mond te sluiten helpen.
Er bestaan medicijnen die het kwijlen minder kunnen maken. Het meest gebruikte medicijn hierdoor is glycopyrrhonium. Soms kan een behandeling van de speekselklieren door middel van botox of door middel van een operatie nodig zijn om er voor zorgen dat kinderen minder kwijlen. Per kind zullen de voor- en nadelen van elke behandeling moeten worden afgewogen.
Verstopping van de darmen
Het medicijn macrogol kan er voor zorgen dat de ontlasting soepel en zacht blijft en stimuleert de darmwand om actief te blijven. Hierdoor kunnen kinderen gemakkelijker hun ontlasting kwijt. erder blijft het belangrijk om te zorgen dat kinderen voldoende vocht en vezels binnen krijgen en zo veel als kan bewegen. Soms zijn zetpillen nodig om de ontlasting op gang te krijgen.

Zindelijkheid
Er kan met zindelijkheidstraining worden begonnen wanneer het kind zelf kan zitten op een potje en interesse begint te krijgen in het potje. Vaak is dit bij kinderen met dit syndroom op latere leeftijd dan gebruikelijk. Tips die kunnen helpen bij het zindelijk worden vindt u in de folder zindelijkheid.
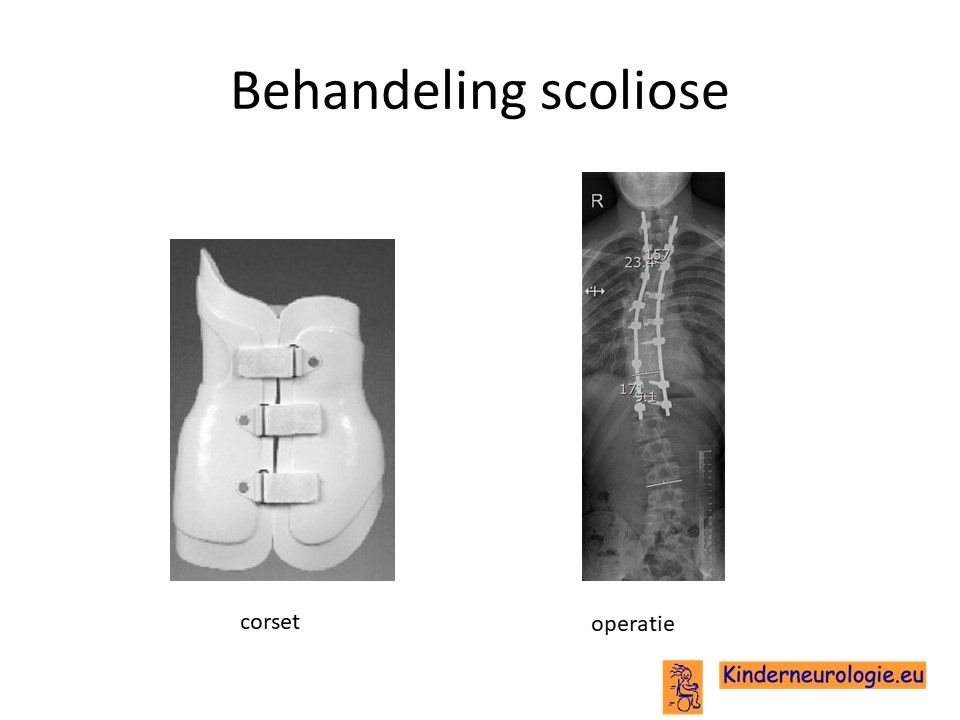
Scoliose
Lichte vormen van verkromming van de wervelkolom kunnen worden behandeld met ene gipscorset om verdergaande verkromming van de wervelkolom te voorkomen. wanneer een gipscorset onvoldoende effect heeft, kan een operatie nodig zijn waarbij de wervels vastgezet. Deze behandeling wordt uitgevoerd door een orthopeed.
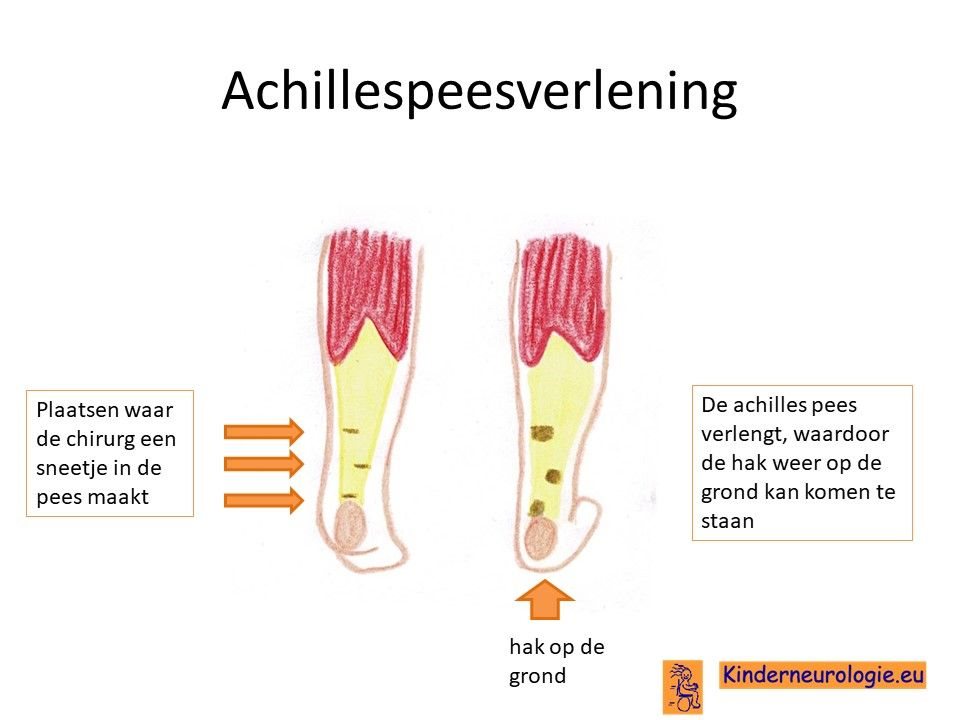
Achillespeesverlenging
Kinderen die slechter gaan lopen als gevolg van tenenloop en hierdoor verkorte achillespezen krijgen, kunnen baat hebben bij een achillespeesverlenging. Ook dit wordt uitgevoerd door een orthopeed.
Pas op met hitte
Kinderen met het Angelman syndroom functioneren vaak minder goed wanneer het heel warm is. Het is belangrijk in de zomer te zorgen voor een koele ruimte waar kinderen kunnen verblijven.

Antibiotica
Een deel van de kinderen die vaak terugkerende infecties heeft, heeft baat bij een lage dosering antibiotica om nieuwe infecties te voorkomen. Per kind moeten de voordelen van het geven van de antibiotica worden afgewogen tegen de nadelen ervan (antibiotica doden ook nuttige bacteriën in de darmen).
Verbeteren botdichtheid
Dagelijks bewegen tijdens daglicht helpt om de botdichtheid te verbeteren. Ook is het belangrijk om voldoende calciumhoudende produkten te eten en gezonde vetten om zelf voldoende vitamine D aan te maken. Wanneer dit niet voldoende is wordt geadviseerd omdagelijks 400IE vitamine D te geven en 500 mg calcium. Soms is het nodig om zogenaamde bisfosfonaten te geven om de botdichtheid te verbeteren.

Onderzoek
Er worden verschillende onderzoeken gedaan om te kijken of er door middel van genetische technieken een mogelijkheid is om er voor te zorgen dat kinderen met het Angelman syndroom kunnen beschikken over meer UBE3A-eiwit. Er wordt gekeken of het chromosoom 15 wat kinderen hebben en van de vader afkomstig is aangezet kan worden tot het maken van het UBE3A-eiwit. Er wordt onderzoek gedaan of het mogelijk is om met zogenaamde anti-sense oligonucleotiden (ASO) te zorgen dat dit vaderlijk chromosoom 15 wordt afgelezen. Daarnaast wordt er onderzoek gedaan of het mogelijk is door middel van een bepaald virus genetische materiaal met daarop het UBE3A-gen in te brengen in hersencellen. Hopelijk is het op deze manier in de toekomst mogelijk om het beloop van het syndroom van Angelman in gunstige zin te beïnvloeden. De onderzoeken met mensen die het Angelman syndroom hebben lopen in het buitenland. In het Europese clinical trial register kan gekeken worden of er momenteel in Europa onderzoeken lopen naar deze aandoening en in het Amerikaanse clinical trial register of dit in Verenigde Staten het geval is.

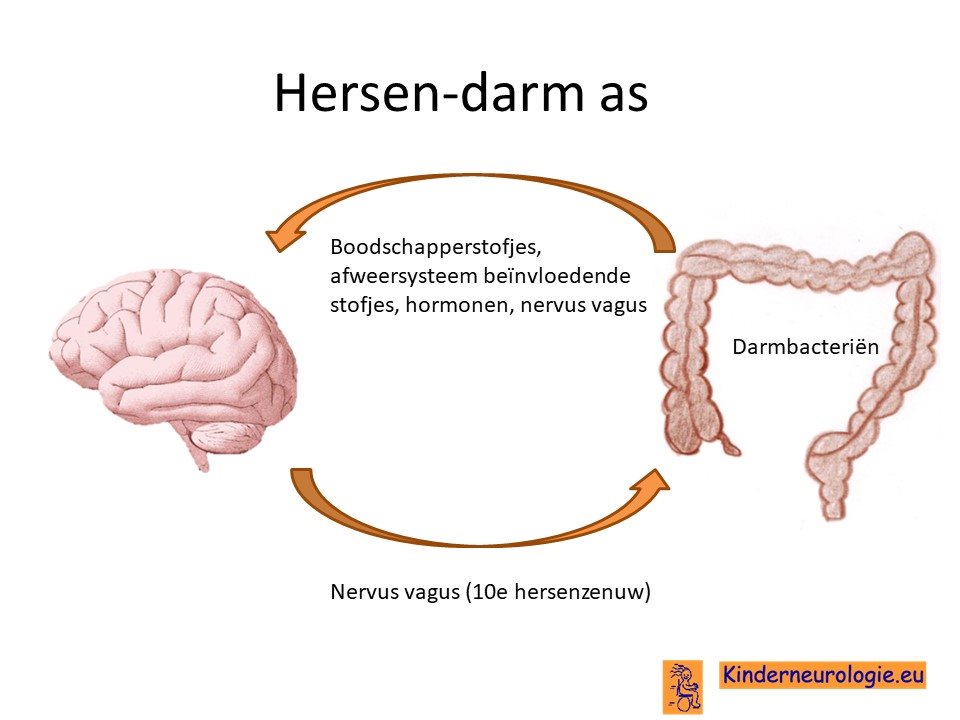
Er wordt ook onderzoek gedaan of beïnvloeden van de hersen-darm-as een gunstig effect kan hebben voor kinderen met het Angelman syndroom. Bij muizen met Angelman syndroom bleek de samenstelling van de darmbacteriën anders te zijn dan bij muizen zonder Angelman syndroom. Muizen die behandeld werden met een linoleenzuur rijk dieet een positieve verandering van de samenstelling in de darmbacteriën kregen en beter gingen functioneren. Mensen zijn echter anders dan muizen, het moet nog onderzocht wordt of een aanpassing van het dieet ook geldt voor mensen met het Angelman syndroom.
Financiële kant van zorg voor een kind met een beperking
De zorg voor een kind met een beperking brengt vaak extra kosten met zich mee. Er bestaan verschillende wetten die zorg voor kinderen met een beperking vergoeden.
Daarnaast bestaan regelingen waar ouders een beroep op kunnen doen, om een tegemoetkoming te krijgen voor deze extra kosten. Meer informatie hierover vindt u in de folder financiën kind met een beperking.

Wat kun je als ouder zelf doen om de ontwikkeling van je kind optimaal te laten verlopen?
Bedenk dat wanneer je samen met je kind speelt, stoeit, danst, zingt, kletst, lacht en/of boekjes leest, dit ook allemaal manieren zijn waarop je kind zijn of haar hersenen traint om stappen voorwaarts te maken in de ontwikkeling. Het is dus niet zo dat alleen momenten van therapie, momenten van training zijn, wat veel ouders denken. Het is daarnaast goed om inspanning af te wisselen met ontspanning, dit is nodig om het geleerde te laten opslaan in de hersenen. De hele dag door training zonder rustmomenten, werkt juist averechts.
Daarnaast is het van onschatbare waarde je kind laten voelen dat je van hem of haar houdt, dat hij/zij geliefd is en zich mag ontwikkelen in een tempo die bij hem of haar past. Dit is extra van belang voor kinderen die zich anders ontwikkelen dan de "norm". "Goed zijn zoals je bent en gesteund te worden door mensen die van je houden is, heel belangrijk voor de ontwikkeling van een kind. Juist de ouders en de andere kinderen in het gezin die dichtbij het kind staan zijn daarin heel belangrijk om het kind daarin dit gevoel te geven. Het is goed dat ouders beseffen wat de waarde hiervan is voor het kind en welke rol zij hierin hebben.
Ook is het belangrijk om te bedenken wat goed voelt voor jullie als gezin en voor jou als ouder en waar jullie energie uithalen. Zorg ervoor dat er bewust ruimte is voor momenten die dit goede gevoel geven. Tot slot is het belangrijk dat je als ouders ook goed voor jezelf zorgt, de zorg voor een kind die zich anders ontwikkelt vraagt nog meer van ouders dan de zorg voor een kind die zich zonder problemen ontwikkelt. Het is goed om voor jezelf te zorgen of te laten zorgen, zodat je als ouder ook de energie houdt, om jouw kind te blijven begeleiden op een manier die bij jou past. Besef dat bij opvoeden hoort om te leren los laten. Veel ouders vinden dit lastig, zeker wanneer hun kind zich anders ontwikkelt dan andere kinderen. Maar dhet kan toch nodig zijn een deel van de zorg op bepaalde momenten uit handen te geven, ook als die ander het anders doet dan jij, je kind leert van deze verschillen en het geeft jou de mogelijk om zelf uit te rusten of nieuwe energie op te doen.

Wat kun je als gezin zelf doen om om te gaan met het hebben van een aandoening bij een gezinslid?
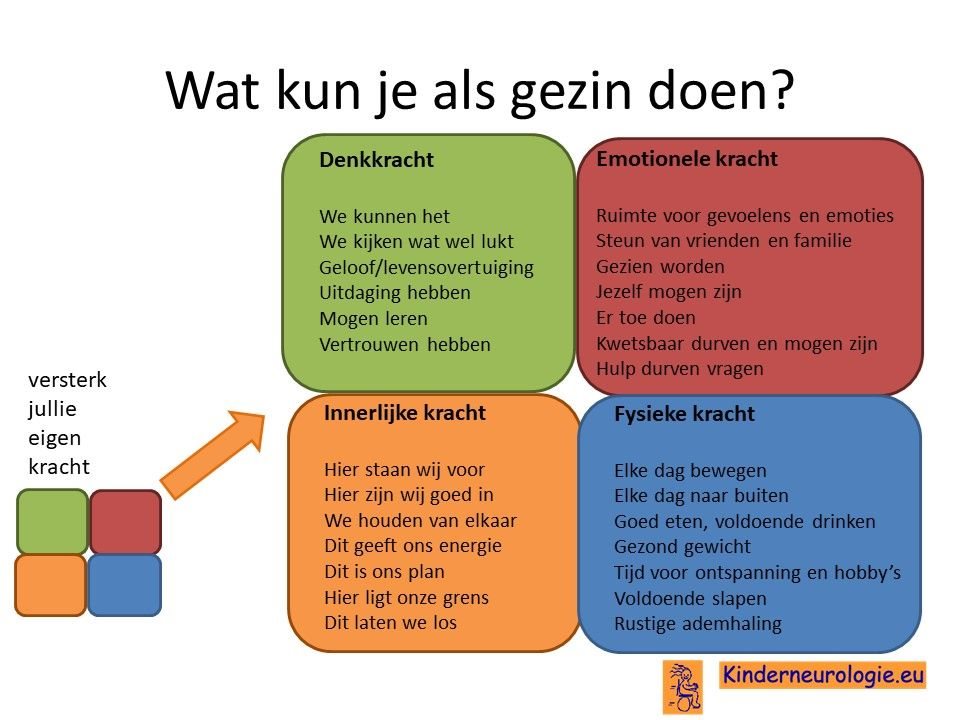
Als gezin van een kind waarbij er sprake is van een aandoening, is het goed om te zorgen dat jullie in de je kracht komen staan. Het is goed om te beseffen over welke denk-, emotionele-, innerlijke- en fysieke kracht jullie als gezin beschikken en hoe jullie deze kracht kunnen inzetten om goed voor ieder lid van het gezin te zorgen. Bekijk wat bij jullie als gezin past. Bekijk wat je kunt doen (of kunt laten) om deze kracht zo optimaal mogelijk in te zetten. En bedenk ook dat ieder lid van het gezin verschillende kwaliteiten heeft waarmee jullie elkaar kunnen aanvullen en kunnen versterken.
Begeleiding
Een maatschappelijk werkende of psycholoog kan begeleiding geven hoe het hebben van deze aandoening een plaatsje kan krijgen in het dagelijks leven. Het kost vaak tijd voor ouders om te verwerken dat de toekomstverwachtingen van hun kind er anders uit zien dan mogelijk verwacht is. Ook vinden veel ouders het vaak lastig hoe zij hun tijd en aandacht moeten verdelen tussen het kind met de beperking en andere kinderen in het gezin. In de folder aandacht en tijd voor brussen vindt u tips die u hierbij kunnen helpen.

Door middel van een oproepje op het forum van deze site kunt u proberen in contact te komen met andere kinderen en hun ouders/verzorgers die ook te maken hebben met het Angelman syndroom. Dit kan ook via de patiëntenvereniging Angelman syndroom.
Wat is de prognose van het Angelman syndroom?
Blijvende problemen
Kinderen die een ontwikkelingsachterstand hebben als gevolg van het Angelman syndroom, blijven deze problemen vaak houden op volwassen leeftijd. De meeste jong volwassenen hebben de hulp van anderen nodig om te kunnen functioneren in het dagelijks leven. De meeste jong volwassenen gaan begeleid wonen.
Ontwikkelingsmogelijkheden
Ouders willen vaak graag weten welke ontwikkelingsmogelijkheden hun kind heeft wanneer er sprake is van een verstandelijke beperking en of hun kind later in staat zal zijn een zelfstandig leven te leiden. Dit is heel moeilijk te voorspellen, zeker wanneer kinderen nog jong zijn, maar ook wanneer kinderen ouder zijn. Een van de factoren die een rol speelt bij de ontwikkelingsmogelijkheden is het IQ van het kind, maar daarnaast spelen ook andere factoren, zoals het wel of niet hebben van epilepsie of ADHD een rol. Het IQ zegt dus zeker niet alles, maar kan wel een bepaalde richting aangeven welke ontwikkelingsmogelijkheden reëel zijn om te verwachten. Dit is overigens wel een gemiddelde verwachting en dat betekent dat er altijd kinderen zijn die deze verwachting zullen behalen, maar dat dit voor anderen toch niet het geval blijkt te zijn of dat er juist meer ontwikkelingsmogelijkheden zijn dan er op grond van het IQ werd verwacht. Het blijft dus belangrijk naar het kind zelf te kijken en te bedenken dat pas op een bepaalde leeftijd duidelijk zal zijn wat het kind uiteindelijk allemaal in staat zal zijn om te leren.
Transitie van zorg
Tussen de leeftijd van 16 en 18 jaar wordt de zorg vaak overgedragen van kinderspecialisten naar specialisten die de zorg aan volwassenen geven. Het is belangrijk om tijdig hierover na te denken. Is er behoefte de zorg over te dragen naar specialisten voor volwassenen of kan de huisarts de zorg leveren die nodig is. En als er behoefte is aan overdragen van de zorg naar specialisten voor volwassenen, naar welke dokter(s) wordt de zorg dan overgedragen? In welk ziekenhuis kan de zorg het beste geleverd worden. Het proces van overdragen van de zorg wordt transitie genoemd. Het is belangrijk hier tijdig over na te denken en een plan voor te maken samen met de dokters die betrokken zijn bij de zorg op de kinderleeftijd.
Ook verandert er veel in de zorg wanneer een jongere de leeftijd van 18 jaar bereikt. Voor meer informatie over deze veranderingen verwijzing wij u naar het artikel veranderingen in de zorg 18+.
![]()
AVG
Een AVG is een arts die zich gespecialiseerd heeft in de zorg voor mensen met een verstandelijke beperking. De AVG richt zich op het voorkomen, behandelen en beperken van lichamelijke en psychische problemen die te maken hebben met een verstandelijke of lichamelijke beperking. De AVG werkt hiervoor samen met de huisarts, de medische specialist, de gedragsdeskundige en/of andere therapeuten (zoals een fysiotherapeut of een logopedist). Er zijn steeds meer poliklinieken in Nederland waar AVG werken en waar kinderen en volwassenen met een verstandelijke beperking terecht kunnen met hun hulpvragen die te maken hebben met hun beperking. Daarnaast werken AVG ook in instellingen en zijn ze betrokken bij gespecialiseerde kinderdagcentra. Op de website van de NVAVG (Nederlandse Vereniging van Artsen voor Verstandelijk Gehandicapten) is een lijst met poliklinieken te vinden.

Epilepsie
Op volwassen leeftijd wordt de ernst van de epilepsie vaak rustiger. Een deel van de volwassenen houdt wel last van epilepsieaanvallen. Na de leeftijd van 40 jaar kan de epilepsie weer toe gaan nemen in ernst. Mogelijk heeft dit met veroudering van de hersenen te maken.
Overgewicht
Volwassenen met het Angelman syndroom hebben vaker last van overgewicht. Overgewicht kan zorgen voor ontstaan van suikerziekte en hart-en vaatziekten. De kans hierop is net zo groot als bij volwassenen met hetzelfde overgewicht zonder het hebben van het Angelman syndroom.
Osteoporose
Volwassenen met het Angelman syndroom hebben vaker last van botontkalking. Dit geldt in het speciaal voor volwassenen die medicijnen gebruiken om epilepsieaanvallen te voorkomen. Het kan nodig zijn om medicatie te geven om de botdichtheid weer te verbeteren (bisfosfonaat).
Relaties
Voor volwassenen met een beperking kan het leggen en behouden van een vriendschap of een relatie met een ander meer moeite kosten dan voor volwassenen zonder beperking. Het gaat minder vanzelfsprekend omdat de volwassene bijvoorbeeld minder energie heeft, het lastiger vindt om zelf contacten te leggen, onzeker is, andere volwassenen niet goed weten hoe met een volwassene met een beperking om te gaan of omdat uitgaansgelegenheden minder goed toegankelijk zijn voor een volwassene met een beperking. Vaak ronden volwassenen hun opleiding af, zodat contact die voorheen via school met klasgenoten plaats vonden, niet meer vanzelfsprekend zijn. Voor een deel van de volwassenen verlopen nieuwe vriendschappen daarna via werk, dagbesteding of de buurt waarin ze wonen. Sport is vaak een mooie manier om nieuwe vriendschappen op te doen. Via de website van uniek sporten, zijn adressen te vinden van sportmogelijkheden voor mensen met een beperking. Ook een hobby kan een mooie manier zijn om nieuwe contacten te leggen. Een ander deel van de volwassen vindt nieuwe vrienden via social media.
Werk
Voor de meeste volwassenen zal het niet mogelijk zijn om regulier werk te vinden. Zij kunnen een beroep doen op de participatiewet. Hiervoor kunnen volwassenen contact opnemen met de gemeente van de plaats waar zij wonen. De gemeente kijkt samen met de volwassene welke ondersteuning de volwassene nodig heeft om passend werk te vinden. Jobcoaches kunnen helpen bij het vinden van passend werk. Een ander deel van de volwassenen gaat naar dagbesteding toe.
Vermoeidheid
Volwassenen met het Angelman syndroom zijn vaak sneller vermoeid dan volwassenen zonder het Angelman syndroom. Dit vraagt vaak aanpassing in het dagelijks leven. Zorgen voor een vast dagritme waarin activiteiten worden afgewisseld met momenten van rust en ontspanning helpt om de energie goed over de dag te verdelen. Ook is het belangrijk elke dag lichamelijk actief te zijn en te zorgen voor een goede conditie. Daarnaast zijn vaste tijden van gaan slapen in een koele donker kamer en vaste tijden van wakker worden belangrijk om te zorgen voor voldoende goede slaap.
Vaak moet er een keuze gemaakt worden welke activiteiten op een dag ingepland gaan worden. Het is goed om te kijken of deze activiteiten noodzakelijk zijn om zelf te doen of niet(wellicht kan iemand anders deze taak overnemen?) en of ze wel of geen energie geven. Op deze manier kan bepaald worden welke activiteiten op een dag het beste ingepland kunnen worden.
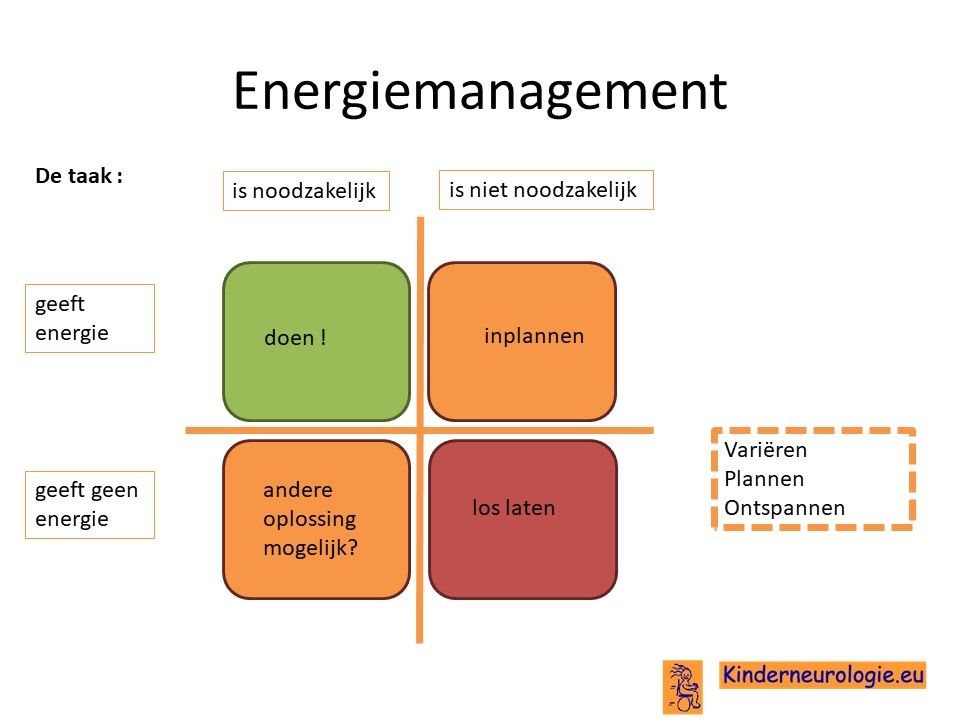
Levensverwachting
De gemiddelde levensverwachting van kinderen met het Angelman syndroom is niet veel anders dan deze van kinderen zonder het Angelman syndroom.
De levensverwachting kan verkort worden in geval van zeer moeilijk behandelbare epilepsie en/of terugkerende longontstekingen waardoor longschade ontstaan is.
Kinderen krijgen
De meeste kinderen die het Angelman syndroom hebben, zullen later zelf als volwassene geen kinderen krijgen. Dit vanwege hun verstandelijke beperking.
Mannen en vrouwen met het Angelman syndroom zijn wel normaal vruchtbaar.
Afhankelijk van de oorzaak van het ontstaan van het Angelman syndroom, bestaat er 50% kans dat deze kinderen ook Angelman syndroom krijgen. Dit geldt vooral voor kinderen met een methylatiestoornis of een translocatie. Een klinisch geneticus kan hier meer informatie over geven. Indien de volwassene geen kinderen wil of kan krijgen, zal nagedacht moeten worden over anticonceptie, waarover u in deze folder meer informatie vindt.
Hebben broertjes en zusjes ook een verhoogde kans om ook het Angelman syndroom te krijgen?
Het Angelman syndroom wordt veroorzaakt door een fout in het erfelijke materiaal van het 15e-chromosoom. Er zijn vier verschillende veranderingen die kunnen zorgen voor het ontstaan van het Angelman syndroom. Elke verandering heeft een eigen kans op herhaling.
Wanneer het Angelman syndroom wordt veroorzaakt door het missen van een stukje van het moederlijk chromosoom 15 (deletie) of doordat beide chromosoom 15 van de vader afkomstig zijn (uniparenterale disomie) dan is de herhalingskans erg laag. Broertjes en zusjes hebben dan minder dan 1% kans om zelf ook het Angelman syndroom te krijgen.
Wanneer er sprake is van een methylatiestoornis dan is de herhalingskans 50%. Broertjes en zusjes hebben dan 50% kans om zelf ook het Angelman syndroom te krijgen.
Wanneer er sprake is van een zogenaamde translocatie, dan is de herhalingskans ongeveer 10%.
Een klinisch geneticus kan hier meer informatie over geven.
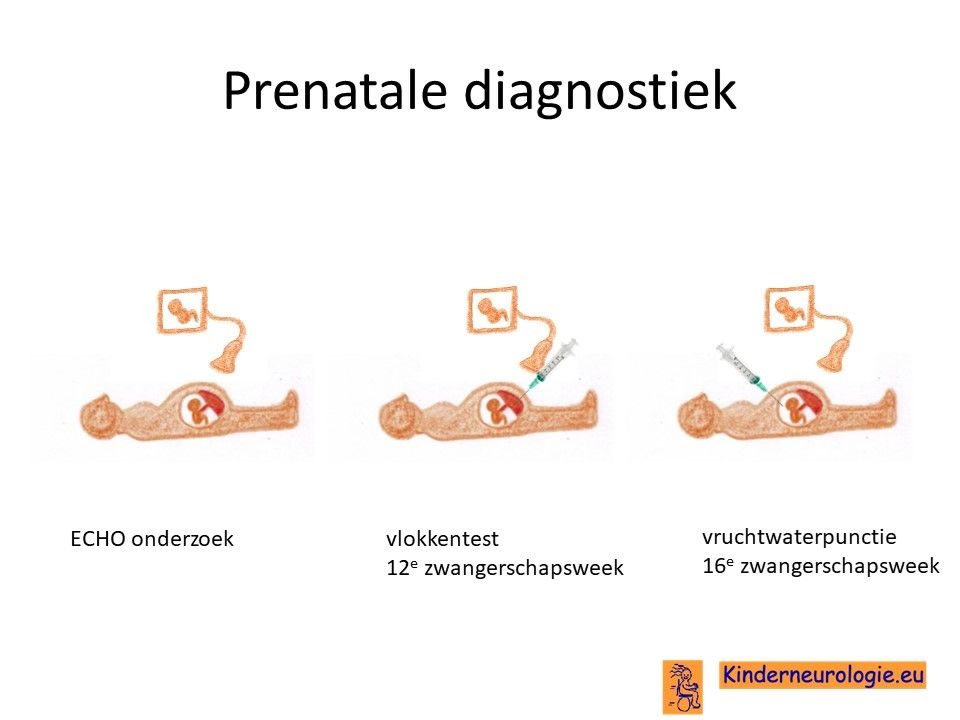
Prenatale diagnostiek
Wanneer bekend is welk foutje in een familie heeft gezorgd voor het ontstaan van het Angelman syndroom, dan is het mogelijk om tijdens een zwangerschap prenatale diagnostiek te verrichten in de vorm van een vlokkentest of een vruchtwaterpunctie om te kijken of dit kindje ook het Angelman syndroom heeft. Beide ingrepen hebben een klein risico op het ontstaan van een miskraam (0,5% bij de vlokkentest en 0,3% bij de vruchtwaterpunctie).De uitslag van deze onderzoeken duurt twee weken. Voor prenatale diagnostiek kan een zwangere de 8ste week verwezen worden door de huisarts of verloskundige naar een afdeling klinische genetica. Meer informatie over prenatale diagnostiek kunt u vinden op de website:www.pns.nl
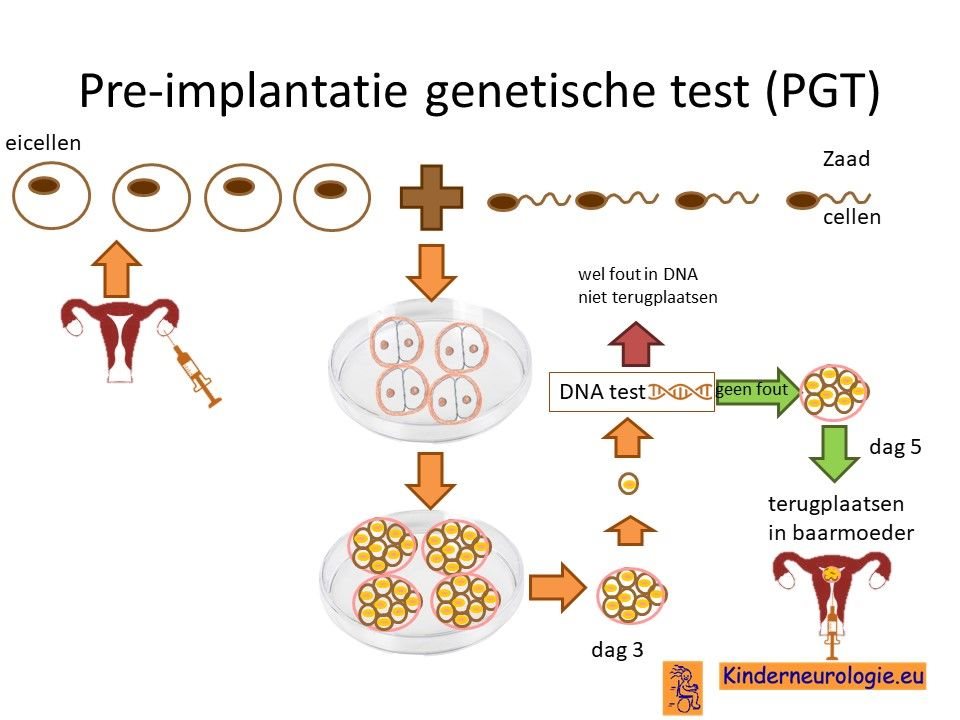
Preïmplantatie Genetische Test (PGT)
Vrouwen die eerder een kindje met het Angelman syndroom hebben gehad als gevolg van een methylatiestoornis of als gevolg van een translocatie kunnen in aanmerking komen voor een preïmplantatie genetische test (PGT.) Bij PGT wordt een vrouw zwanger door middel van IVF (In Vitro Fertilisatie). De bevruchting vindt dan buiten het lichaam plaats, waardoor het zo ontstane pre-embryo onderzocht kan worden op het hebben van het Angelman-syndroom. Alleen embryo’s zonder de aanleg voor Angelman-syndroom, komen in aanmerking voor terugplaatsing in de baarmoeder. Voor meer informatie zie www.pgtnederland.nl.
Wilt u dit document printen dan kunt u hiervoor de printbutton bovenaan de pagina gebruiken
Wilt u ook uw verhaal kwijt, dat kan: verhalen kunnen gemaild worden via via contact en zullen daarna zo spoedig mogelijk op de site worden geplaatst. Voor meer informatie zie hier.
Heeft u foto's die bepaalde kenmerken van deze aandoening duidelijk maken en die hier op de website mogen worden geplaatst, dan vernemen wij dit graag via contact.
Links
www.angelmansyndroom.nl
(site van de Angelman vereniging)
Referenties
- Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, Wagstaff J. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006;140:413-8.
- Valente KD, Koiffmann CP, Fridman C, Varella M, Kok F, Andrade JQ, Grossmann RM, Marques-Dias MJ. Epilepsy in patients with angelman syndrome caused by deletion of the chromosome 15q11-13. Arch Neurol. 2006;63:122-8.
- Neurologic manifestations of Angelman syndrome. Thibert RL, Larson AM, Hsieh DT, Raby AR, Thiele EA. Pediatr Neurol. 2013;48:271-9.
- Angelman syndrome: Current and emerging therapies in 2016. Tan WH, Bird LM. Am J Med Genet C Semin Med Genet. 2016;172:384-401
- Sleep in Angelman syndrome: A review of evidence. Spruyt K, Braam W, Curfs LM. Sleep Med Rev. 2018;37:69-84.
- Quantitative proteomics reveals neuronal ubiquitination of Rngo/Ddi1 and several proteasomal subunits by Ube3a, accounting for the complexity of Angelman syndrome. Ramirez J, Lectez B, Osinalde N, Sivá M, Elu N, Aloria K, Procházková M, Perez C, Martínez-Hernández J, Barrio R, Šašková KG, Arizmendi JM, Mayor U. Hum Mol Genet. 2018 ;27:1955-1971
- An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Bindels-de Heus KGCB, Mous SE, Ten Hooven-Radstaake M, van Iperen-Kolk BM, Navis C, Rietman AB, Ten Hoopen LW, Brooks AS; ENCORE Expertise Center for AS, Elgersma Y, Moll HA, de Wit MY. Am J Med Genet A. 2020;182:53-63
- Epilepsy in Angelman syndrome: A scoping review. Samanta D. Brain Dev. 2021;43:32-44
- Clinical aspects of a large group of adults with Angelman syndrome. den Besten I, de Jong RF, Geerts-Haages A, Bruggenwirth HT, Koopmans M; ENCORE Expertise Center for AS 18+, Brooks A, Elgersma Y, Festen DAM, Valstar MJ. Am J Med Genet A. 2021;185:168-181
- A multidisciplinary approach and consensus statement to establish standards of care for Angelman syndrome. Duis J, Nespeca M, Summers J, Bird L, Bindels-de Heus KGCB, Valstar MJ, de Wit MY, Navis C, Ten Hooven-Radstaake M, van Iperen-Kolk BM, Ernst S, Dendrinos M, Katz T, Diaz-Medina G, Katyayan A, Nangia S, Thibert R, Glaze D, Keary C, Pelc K, Simon N, Sadhwani A, Heussler H, Wheeler A, Woeber C, DeRamus M, Thomas A, Kertcher E, DeValk L, Kalemeris K, Arps K, Baym C, Harris N, Gorham JP, Bohnsack BL, Chambers RC, Harris S, Chambers HG, Okoniewski K, Jalazo ER, Berent A, Bacino CA, Williams C, Anderson A. Mol Genet Genomic Med. 2022;10:e1843
- From first report to clinical trials: a bibliometric overview and visualization of the development of Angelman syndrome research. Zampeta FI, Distel B, Elgersma Y, Iping R. Hum Genet. 2022;141:1837-1848
- Prader-Willi and Angelman Syndromes: Mechanisms and Management. Ma VK, Mao R, Toth JN, Fulmer ML, Egense AS, Shankar SP. Appl Clin Genet. 2023;16:41-52.
- Sleep disturbance in Angelman syndrome patients. Qu S, Wang J, Guan X, Song C, Wang Y. Orphanet J Rare Dis. 2024;19:146
- Gut microbes in central nervous system development and related disorders. Gan Y, Chen Y, Zhong H, Liu Z, Geng J, Wang H, Wang W. Front Immunol. 2024;14:1288256.
-
Precision Medicine in Angelman Syndrome. Manssen L, Krey I, Gburek-Augustat J, von Hagen C, Lemke JR, Merkenschlager A, Weigand H, Makowski C. Neuropediatrics. 2025;56:69-82
Laatst bijgewerkt: 30 juli 2025 voorheen: 14 augustus 2024, 29 juni 2023, 8 juni 2022 , 21 april 2021,31 maart 2021, 18 maart 2020, 18 januari 2020, 20 februari 2019, 18 juli 2018 en 17 september 2007
auteur: JH Schieving