Wat is het O'Donnell-Luria-Rodan syndroom?
Het O'Donnell-Luria-Rodan syndroom is een syndroom waarbij kinderen een algehele ontwikkelingsachterstand hebben al dan niet in combinatie met een of meerdere bijzondere uiterlijke kenmerken.
Hoe wordt het O'Donnell-Luria-Rodan syndroom ook wel genoemd?
Het O'Donnell-Luria-Rodan syndroom is genoemd naar twee artsen O'Donnell-Luria en Rodan die dit syndroom beschreven hebben. Het wordt afgekort met de letters ODLURO.
KMT2E-gerelateerd syndroom
Een andere naam die ook gebruikt wordt is het KMT2E-gerelateerd syndroom. KMT2E is de naam van de plaats in het erfelijk materiaal (het DNA) waar kinderen met dit syndroom een fout hebben zitten die verantwoordelijk is voor het ontstaan van de symptomen die horen bij dit syndroom. Door deze naam te gebruiken is het voor iedereen duidelijk dat er sprake is van deze fout. Ook wordt KMT2E-syndroom als naam gebruikt.
Hoe vaak komt het O'Donnell-Luria-Rodan syndroom voor?
Het O'Donnell-Luria-Rodan syndroom is een zeldzame ziekte. Het is niet precies bekend hoe vaak het O'Donnell-Luria-Rodan syndroom voorkomt.
Waarschijnlijk is bij een deel van de kinderen die het O'Donnell-Luria-Rodansyndroom heeft, de juiste diagnose ook niet gesteld, omdat het syndroom niet herkend is. Dit syndroom is sinds 2019 bekend als syndroom.
Door nieuwe genetische technieken zoals exome sequencing zal deze diagnose waarschijnlijk vaker gesteld gaan worden bij kinderen en volwassenen met dit syndroom.

Bij wie komt het O'Donnell-Luria-Rodan syndroom voor?
Het O'Donnell-Luria-Rodan syndroom is al voor de geboorte aanwezig. Het kan wel enige tijd duren voordat duidelijk is dat er sprake is van het O'Donnell-Luria-Rodan syndroom.
Zowel jongens als meisjes kunnen het O'Donnell-Luria-Rodan syndroom krijgen. Toch wordt het syndroom vaker gezien bij jongens dan bij meisjes zonder dat bekend is waarom.
Waar wordt het O'Donnell-Luria-Rodan syndroom door veroorzaakt?
Fout in het erfelijk materiaal
Het O'Donnell-Luria-Rodansyndroom wordt veroorzaakt door een fout op een stukje materiaal op het 7e-chromosoom. De plaats van dit foutje werd het KMT2E-gen genoemd.
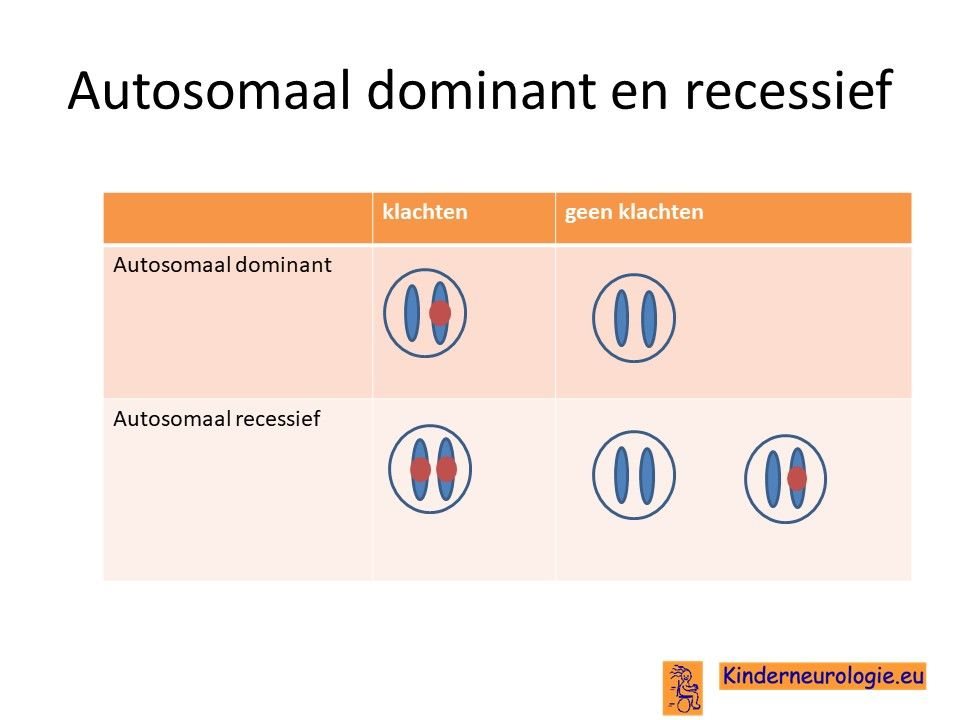
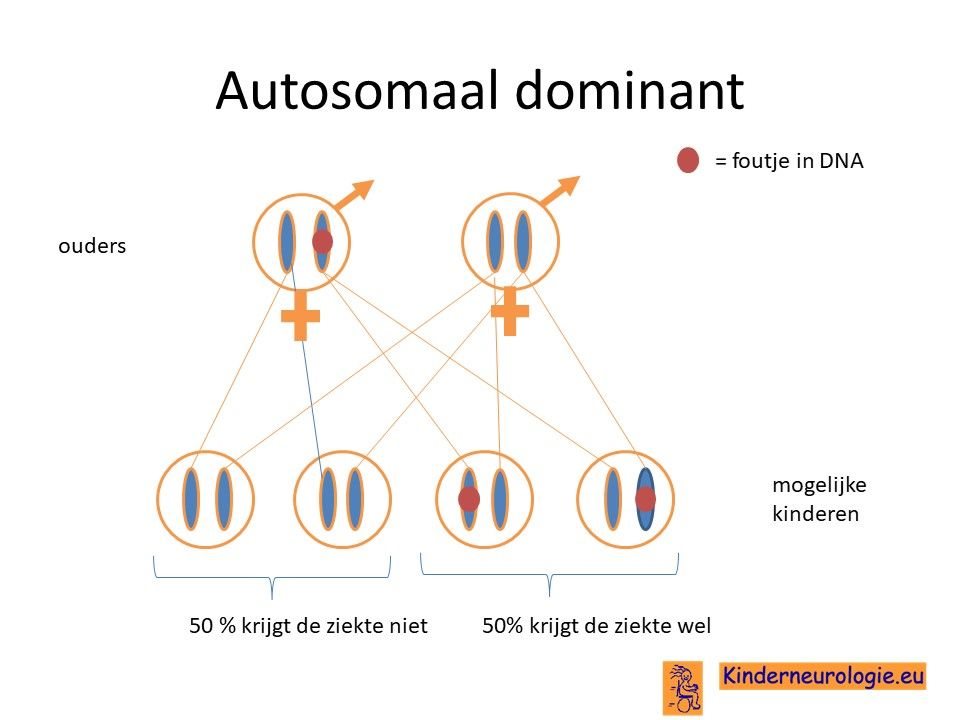
Autosomaal dominant
Het O'Donnell-Luria-Rodan syndroom wordt veroorzaakt door een zogenaamde autosomaal dominant fout. Dit houdt in dat een fout op een van de twee chromosomen 7 die een kind heeft in het KMT2E-gen al voldoende is om de aandoening te krijgen. Dit in tegenstelling tot een autosomaal recessief fout waarbij kinderen pas klachten krijgen wanneer beide chromosomen 7 een fout bevatten.
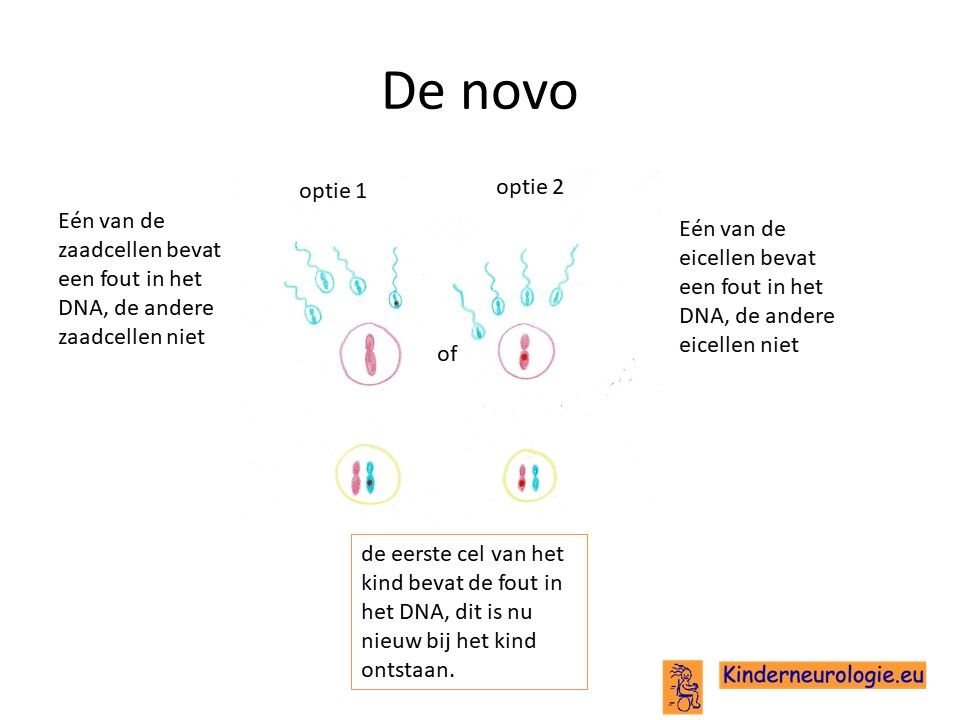
Bij het kind zelf ontstaan
Bij het merendeel van de kinderen met een O'Donnell-Luria-Rodansyndroom is de fout bij het kind zelf ontstaan na de bevruchting van de eicel door de zaadcel en niet overgeërfd van een van de ouders.
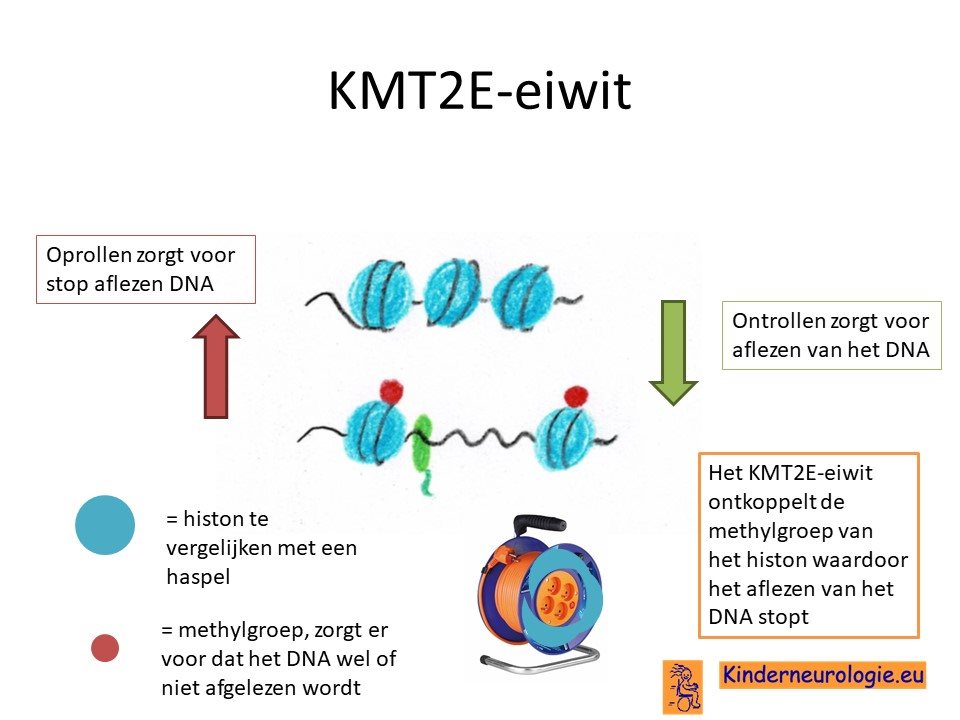
Afwijkend eiwit
Als gevolg van een van bovengenoemde foutjes in het erfelijk materiaal worden een bepaald eiwit niet goed worden gemaakt. Dit eiwit heet lysine-specifiek methyltransferase 2E ook wel afgekort als KMT2E. Dit eiwit speelt een belangrijke rol bij het hangen van een methylgroep aan histonen. Deze histonen zijn te vergelijken met een haspel waar het DNA mee kan worden opgerold of worden afgerold. Het DNA moet op het juiste moment tijdens de ontwikkeling van de hersenen worden afgerold om te kunnen worden afgelezen. Op andere momenten moeten het DNA weer worden opgerold, wanneer het niet hoeft te worden afgelezen. Wanneer het KMT2E-eiwit niet goed werkt, dan verloopt dit op- en afrollen van het DNA niet goed waardoor het DNA niet op de juiste momenten wordt afgelezen. Hierdoor worden de hersenen anders aangelegd dan gebruikelijk.
Netwerken
In de hersenen bevinden zich allerlei netwerken van hersencellen die nodig zijn om een bepaalde taak goed te kunnen uitvoeren. Door de fout in het KMT2E-gen worden deze netwerken minder goed aangelegd en kunnen de hersencellen minder goed met elkaar samen werken. Dit veroorzaakt de symptomen die horen bij dit syndroom.
Wat zijn de symptomen van het O'Donnell-Luria-Rodan syndroom?
Variatie
Er bestaat een grote variatie in de hoeveelheid en de ernst van de symptomen die verschillende kinderen met het O'Donnell-Luria-Rodan syndroom hebben.
Dit valt van te voren niet goed te voorspellen van welke symptomen een kind last zal krijgen. Geen kind zal alle onderstaande symptomen tegelijkertijd hebben. Dat betekent dat onderstaande kenmerken kunnen voorkomen, maar niet hoeven voor te komen.

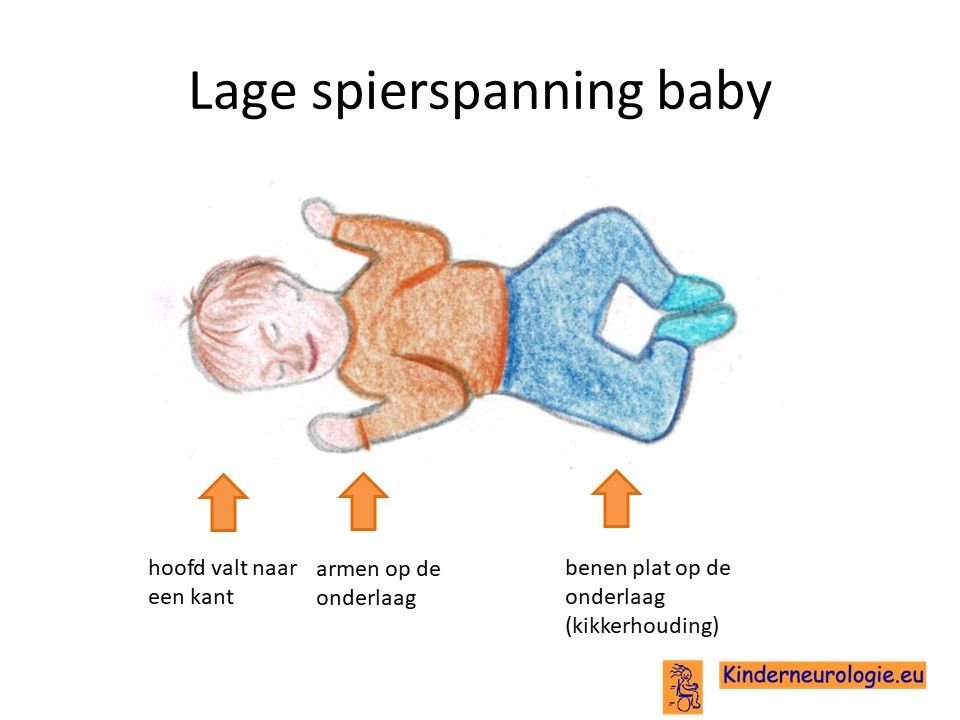
Lage spierspanning
Kinderen met het O'Donnell-Luria-Rodan syndroom hebben vaak een lagere spierspanning. Hierdoor voelen ze slapper aan dan andere kinderen. Wanneer kinderen opgetild worden, moeten ze goed ondersteund worden. Door de lagere spierspanning kunnen bijvoorbeeld de gewrichten van de ellebogen en de knieën gemakkelijk overstrekt worden. Dit wordt hypermobiliteit genoemd. Veel kinderen met het O'Donnell-Luria-Rodan syndroom hebben platvoetjes door de lagere spierspanning.
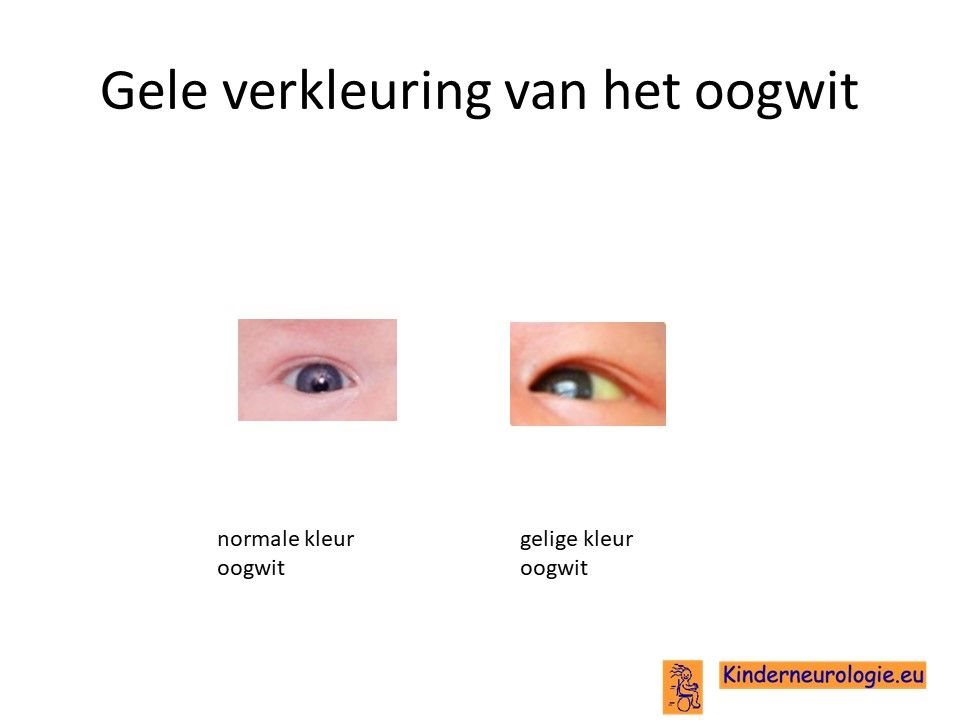
Geelzien
Jonge baby's met dit syndroom kunnen na de geboorte een gele kleur van het oogwit en de huid krijgen.
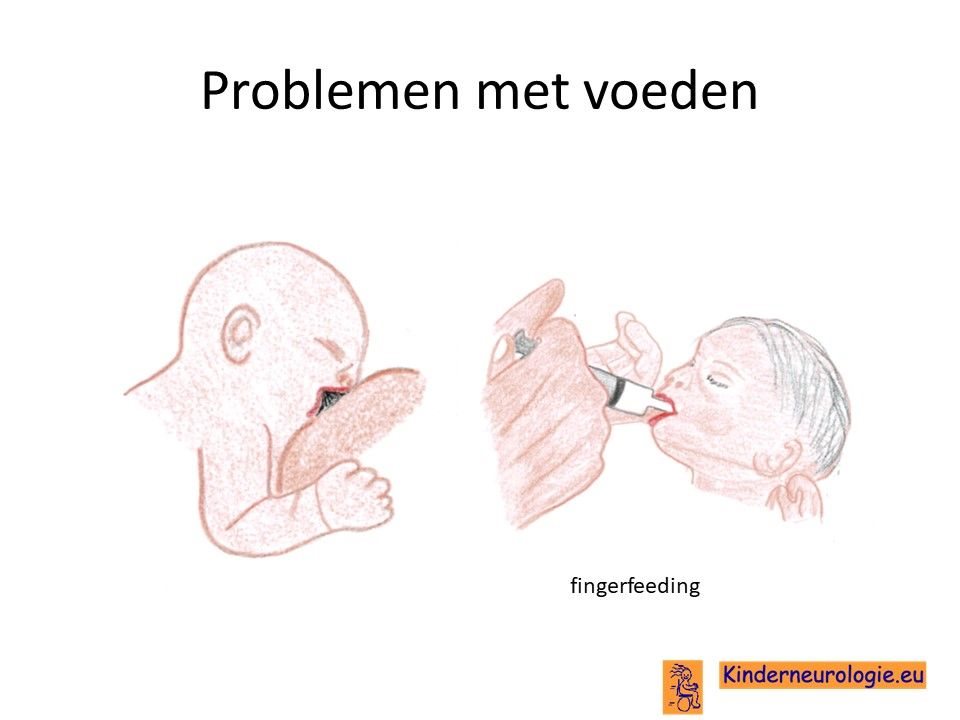
Problemen met drinken
Een groot deel van de baby’s met het O'Donnell-Luria-Rodansyndroom heeft problemen met drinken. Ze drinken langzaam en laten de borst of speen vaak los. Het kost vaak veel tijd om baby’s met dit syndroom de borst of de fles te geven. Ook spugen kinderen met dit syndroom gemakkelijk. Met het ouder worden, verloopt het drinken bij de meeste kinderen wel beter.
Licht van gewicht
Door de problemen met drinken blijven kinderen met dit syndroom in het eerste levensjaar vaak licht van gewicht. Ze groeien minder hard dan hun leeftijdsgenoten. Dit wordt ook wel failure to thrive genoemd.
Ontwikkelingsachterstand
Kinderen met het O'Donnell-Luria-Rodansyndroom ontwikkelen zich langzamer dan hun leeftijdsgenoten. Ze gaan later rollen, zitten en staan dan hun leeftijdsgenoten. Het lopen verloopt vaak wat houterig, kinderen kunnen gemakkelijker vallen. De meeste kinderen leren al deze vaardigheden wel, maar op een latere leeftijd dan hun leeftijdsgenoten. Vaak hebben kinderen met dit syndroom problemen met de fijne motoriek, zoals met schrijven, tekenen of knippen. Dit is voor hen veel lastiger dan voor leeftijdsgenoten. Dit wordt dyspraxie genoemd.
Problemen met praten
Voor veel kinderen met het O'Donnell-Luria-Rodansyndroom is het moeilijker om te leren praten. De eerste woordjes komen vaak later dan gebruikelijk. . De meeste kinderen hebben op oudere leeftijd een beperktere woordenschat en vinden het lastiger om goedlopende zinnen te maken. De zinnen die ze maken zijn vaak kort en bevatten weinig woorden. Het begrijpen van taal van anderen gaat kinderen met dit syndroom vaak beter af dan het zelf spreken.
Kinderen met dit syndroom hebben ook vaak een lagere spierspanning in het gezicht, waardoor ze de woorden en zinnen minder duidelijk uitspreken en voor onbekende soms moeilijker verstaanbaar zijn. Dit wordt dysarthrie genoemd.
Problemen met leren
Kinderen met het O'Donnell-Luria-Rodansyndroom hebben bijna allemaal problemen met leren. De mate van problemen met leren verschilt, sommige kinderen kunnen naar regulier onderwijs, andere zijn moeilijk lerend of zeer moeilijk lerend. Bij een deel van de kinderen is het IQ lager dan 70, de grens waaronder wordt gesproken van een verstandelijke beperking.
Open mond
Vaak hebben de spieren in het gezicht weinig spierspanning, waardoor kinderen gemakkelijk hun mond open hebben. Door de lager spierspanning rondom de mond hebben kinderen ook meer moeite om duidelijk verstaanbaar te praten. Ook kan de open mond zorgen voor het ontstaan van kwijlen.

AD(H)D
AD(H)D komt vaker voor bij kinderen met dit syndroom. Kinderen met ADHD hebben moeite om bij een taakje langere tijd de aandacht te houden. Ze spelen maar kort met een bepaald speelgoed en gaan dan weer naar een ander stukje speelgoed. Kinderen zijn snel afgeleid door een geluid of een beweging in de kamer. Op school hebben kinderen moeite langer tijd hun aandacht bij het schoolwerk te houden.
Kinderen kunnen moeite hebben met stil zitten en bewegen het liefst de hele dag. Kinderen hebben de neiging om eerst te doen en dan pas te denken of dit wel verstandig is, dit wordt impulsief gedrag genoemd.
Autistiforme kenmerken
Kinderen met een O'Donnell-Luria-Rodansyndroom hebben vaker autistiforme kenmerken. Ze zijn meer in zich zelf gekeerd en hebben niet zo’n behoefte aan contact met andere mensen.
Kinderen met autistiforme kenmerken houden vaak van een vaste voorspelbare structuur in de dag en vinden het lastig wanneer hiervan af geweken wordt of wanneer er onverwachte gebeurtenissen zijn. Kinderen kunnen door onverwachte gebeurtenissen heel boos of juist heel verdrietig worden, omdat ze niet goed weten hoe ze hier mee om moeten gaan.
Ook hebben kinderen vaak voorkeur voor bepaald speelgoed of een bepaalde hobby waar ze zich heel lang mee kunnen vermaken.

Stereotypieën
Veel kinderen maken graag bewegingen met hun armen en hun handen die vaak terug keren. Zulke bewegingen worden stereotypieën genoemd. Sommige kinderen gaan wapperen met hun handen, anderen maken draaiende bewegingen of wrijvende bewegingen over de borst heen. Deze bewegingen komen vaak voor wanneer kinderen iets heel leuks of iets spannends gaan doen.

Gemakkelijk boos worden
Kinderen met dit syndroom kunnen gemakkelijk boos worden. Vaak is er wel een aanleiding voor het ontstaan van deze boosheid, bijvoorbeeld omdat iets niet lukt of omdat kinderen iets niet mogen, maar lang niet altijd is dit duidelijk. Kinderen kunnen in hun boosheid andere mensen of zich zelf pijn doen door te knijpen of te bijten. Kinderen vinden het moeilijk om zelf uit deze boosheidsaanval te komen. Vaak hebben ze daarbij de hulp van anderen nodig.
Skin-picking
Een deel van de kinderen plukt aan de eigen huid, waardoor wondjes van de huid kunnen ontstaan. Dit wordt skin (Engelse woord voor huid) picking (Engelse woord voor plukken genoemd).
Angst
Kinderen met dit syndroom hebben gemakkelijker last van angsten. Bijvoorbeeld angst om alleen zonder de ouders te zijn, angst voor het donker of angst voor onbekende en vreemde situaties.
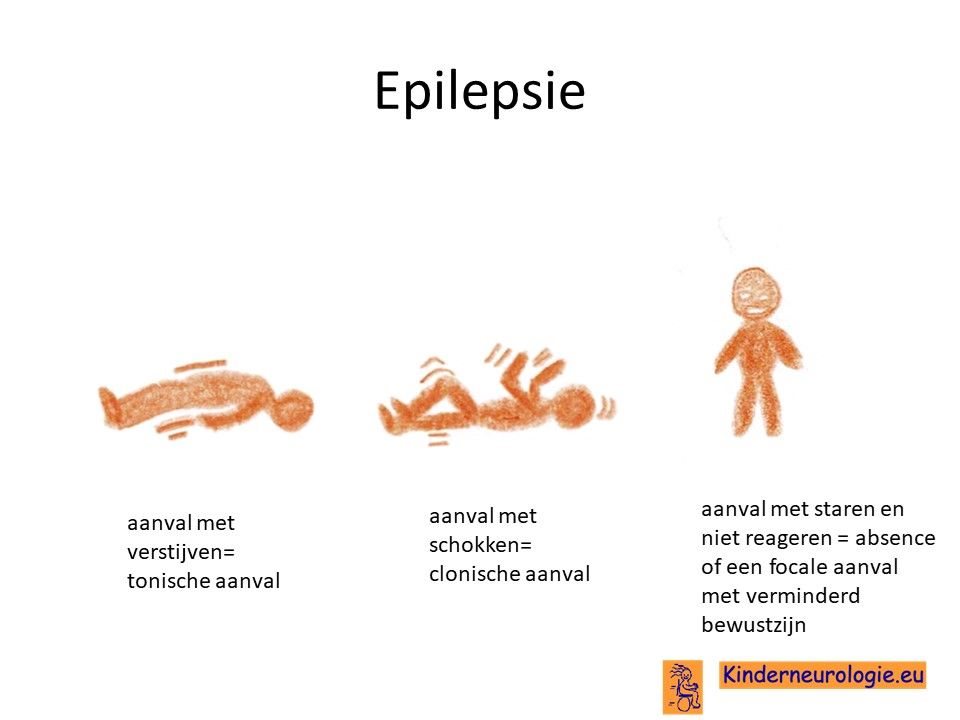
Epilepsie
Een deel van de kinderen met het O'Donnell-Luria-Rodan syndroom (geschat 15%) krijgt last van epilepsie aanvallen. Verschillende type epilepsie aanvallen kunnen voorkomen. Veel voorkomende aanvallen zijn aanvallen met verstijven (tonische aanvallen genoemd), aanvallen met herhaalde schokken (clonische aanvallen genoemd), aanvallen met kleine schokjes op verschillende plaatsen in het lichaam (myoclonieën) of aanvallen met staren (focale aanvallen met verminderd bewustzijn genoemd). Ook komen koortsstuipen vaker voor bij kinderen met dit syndroom. Epilepsie blijkt vaker voor te komen bij meisjes dan bij jongens.
Problemen met slapen
Slaapproblemen komen vaak voor bij kinderen met dit syndroom. Veel kinderen vinden het lastig om in de avond in slaap te vallen. Kinderen kunnen licht slapen en gemakkelijk wakker worden door geluiden in de omgeving. Sommige kinderen worden heel vroeg wakker. Bij een deel van de kinderen worden de slaapproblemen veroorzaakt door epilepsie gedurende de nacht.

Uiterlijke kenmerken
Bij veel syndromen hebben kinderen vaak wat veranderde uiterlijke kenmerken. Hier hebben kinderen zelf geen last van, maar het kan de dokters helpen om te herkennen dat er sprake is van een syndroom en mogelijk ook van welk syndroom. Ook maakt dit vaak dat kinderen met hetzelfde syndroom vaak meer op elkaar lijken dan op hun eigen broertjes en zusjes, terwijl de kinderen toch niet familie van elkaar zijn.
Kinderen met het O'Donnell-Luria-Rodan syndroom hebben vaak een groot hoofd die een langwerpige vorm heeft. Het voorhoofd is vaak hoog. De ogen kunnen diep liggen doordat de huid rondom de ogen wat gezwollen is. Vaak lopen de ogen in de richting van de oren een stukje omlaag. De wangen zijn vaak bol, waardoor de groef tussen de neus en de wangen diep is.
Groot hoofd
Kinderen met dit syndroom groeit het hoofd vaak sneller dan van leeftijdsgenoten. Bij een deel van de kinderen is het hoofd veel groter dan gebruikelijk. Een veel te groot hoofd wordt macrocefalie genoemd.
Handen
Vaak worden de vingers van de handen smaller toe in de richting van de vingertoppen.
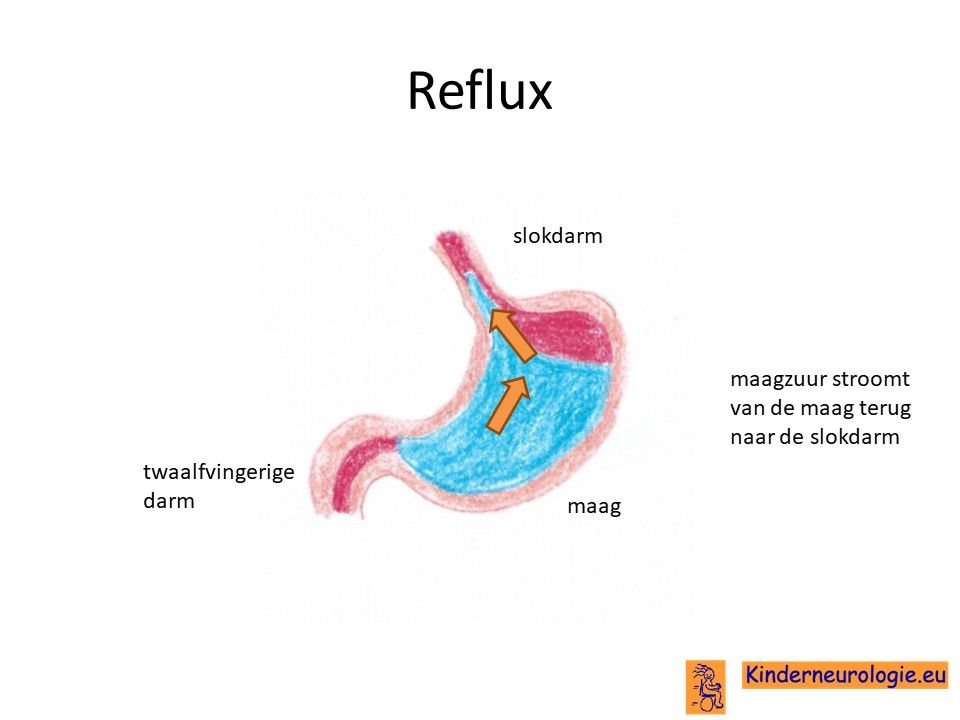
Reflux
Kinderen met het O'Donnell-Luria-Rodan syndroom hebben heel vaak last van het terugstromen van voeding vanuit de maag naar de slokdarm. Dit wordt reflux genoemd. Omdat de maaginhoud zuur is, komt het zuur zo ook in de slokdarm, soms zelfs ook in de mond. Dit zuur kan zorgen voor pijnklachten, waardoor kinderen moeten huilen en soms ook niet willen eten. Ook kan het maken dat kinderen moeten spugen.
Door het zuur kan de slokdarm geïrriteerd en ontstoken raken. Wanneer dit niet tijdige ontdekt en behandeld wordt, kan dit zorgen voor het spuug met daarin bloedsliertjes.
Kwijlen
Kinderen met dit syndroom hebben gemakkelijk last van kwijlen. Dit komt door slapheid van de spieren in het gezicht en rondom de mond, waardoor het speeksel gemakkelijk uit de mond loopt.
Verstopping van de darmen
Verstopping van de darmen komt vaak voor bij kinderen met het O'Donnell-Luria-Rodan syndroom. De ontlasting komt dan niet elke dag en is vaak hard waardoor kinderen moeite hebben met poepen.
Zindelijkheid
De meeste kinderen met dit syndroom worden op latere leeftijd zindelijk dan gebruikelijk.

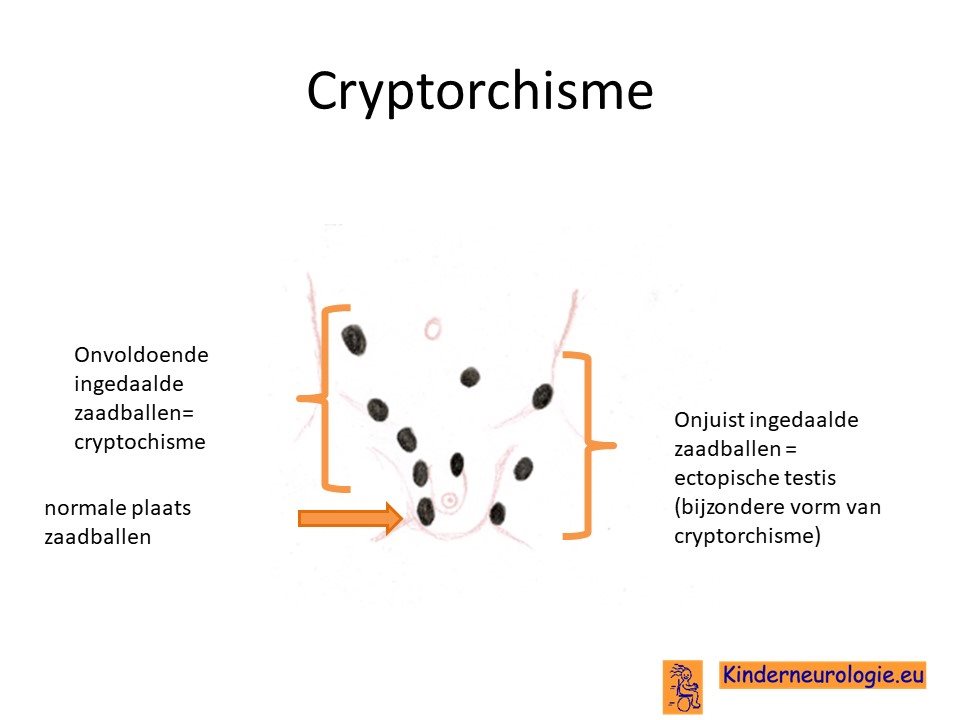
Niet ingedaalde zaadballen
Bij een deel van de jongens zitten de zaadballen niet in de balzak. Dit wordt cryptorchisme genoemd.
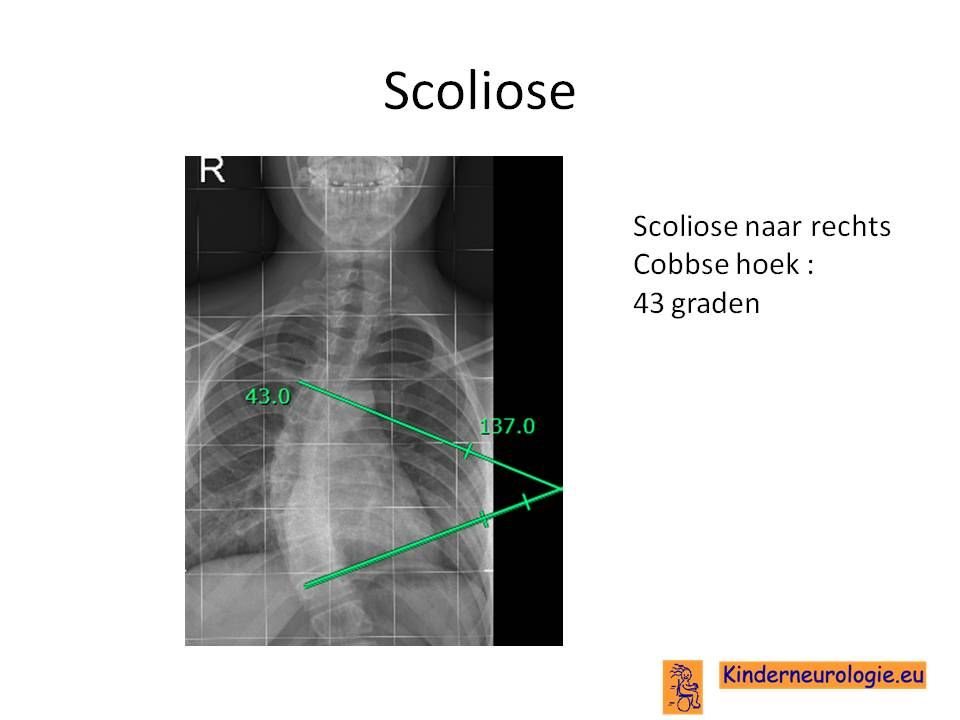
Scoliose
Een deel van de kinderen met het O'Donnell-Luria-Rodan syndroom krijgt een zijwaartse verkromming van de rug. Dit wordt een scoliose genoemd. Ook komt een toegenomen kromming van de bovenrug vaker voor, waardoor kinderen voorover gebogen lopen. Dit wordt kyfose genoemd.
Hoe wordt de diagnose O'Donnell-Luria-Rodan syndroom gesteld?
Verhaal en onderzoek
Op grond van het verhaal van een kind met een ontwikkelingsachterstand en enkele opvallende uiterlijke kenmerken kan vermoed worden dat er sprake is van een syndroom. Er zijn echter veel verschillende syndromen die allemaal voor deze symptomen kunnen zorgen. Vaak zal aanvullend onderzoek nodig zijn om aan de diagnose O'Donnell-Luria-Rodan syndroom te stellen.
Bloedonderzoek
Bij routine bloedonderzoek worden bij kinderen met het O'Donnell-Luria-Rodan syndroom geen bijzonderheden gevonden.
Genetisch onderzoek
Wanneer aan de diagnose gedacht wordt, kan door middel van gericht genetisch onderzoek op bloed naar het voorkomen van een foutje op het 7e-chromosoom in het KMT2E-gen
Vaak worden ook alle chromosomen tegelijkertijd onderzocht (zogenaamd Array onderzoek), soms kan op deze manier de diagnose O'Donnell-Luria-Rodan syndroom worden gesteld omdat een stuk van chromosoom 7 mist waarop het KMT2E-gen ligt.
In de toekomst zal door middel van een nieuwe genetische techniek (exome sequencing genoemd) mogelijk ook deze diagnose gesteld kunnen worden zonder dat er specifiek aan gedacht was of naar gezocht is.
MRI-scan
Bij kinderen met een ontwikkelingsachterstand zal vaak een MRI scan gemaakt worden om te kijken of er bijzonderheden aan de hersenen te zien zijn. Bij een groot deel van de kinderen ziet deze MRI-scan er helemaal normaal uit. Bij een deel van de kinderen worden wel afwijkingen gezien, maar deze afwijkingen komen ook voor bij kinderen met andere syndromen en zijn niet specifiek voor het O'Donnell-Luria-Rodansyndroom. Afwijkingen die vaker voorkomen zijn een andere vorm van de hersenbalk, een te witte kleur van de zogenaamde witte stof omdat het myelinelaagje anders en later wordt aangelegd (vertraagd myelinisatie). Soms worden kleine cystes in de hersenen gezien.
Stofwisselingsonderzoek
Kinderen met een ontwikkelingsachterstand krijgen vaak stofwisselingsonderzoek van bloed en urine om te kijken of er sprake is van een stofwisselingsziekte die verklarend is voor de ontwikkelingsachterstand. Bij kinderen met het O'Donnell-Luria-Rodan syndroom worden hierbij geen bijzonderheden gezien.
EEG
Kinderen met epilepsie krijgen vaak een EEG om te kijken van welk soort epilepsie er sprake is. Op het EEG worden vaak epileptiforme afwijkingen gezien. Deze afwijkingen zijn niet kenmerkend voor het O'Donnell-Luria-Rodansyndroom, maar kunnen bij veel andere syndromen met epilepsie ook gezien worden.
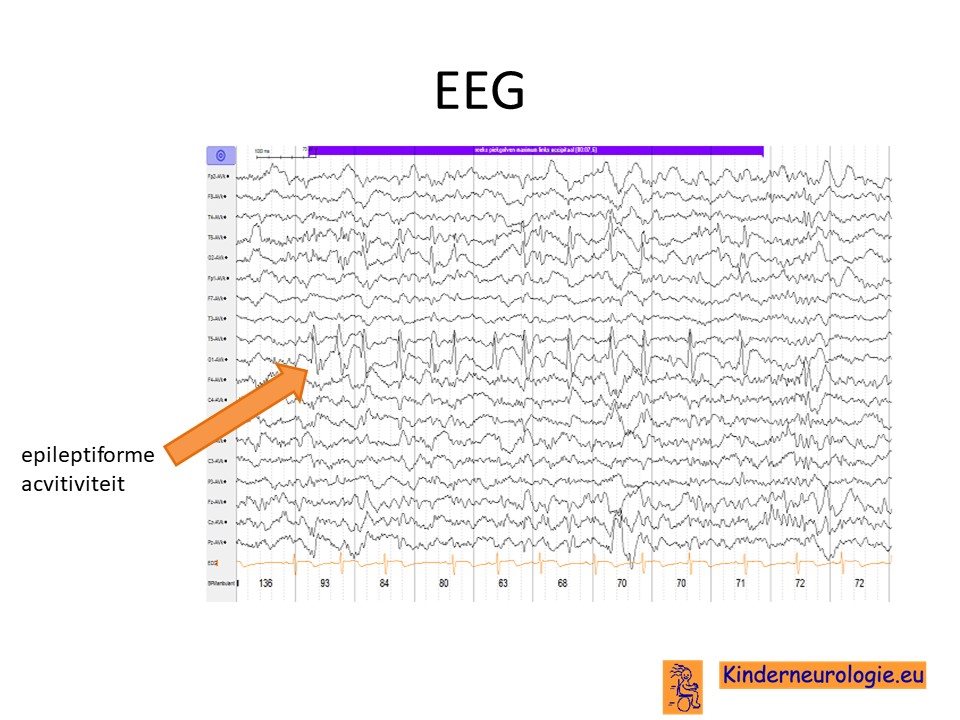
Foto van de rug
Op een foto kan te zien zijn dat de botleeftijd minder verder is dan gebruikelijk voor de leeftijd van het kind. Ook kan een foto te zien zijn in welke mate er sprake is van een zijwaartse verkromming van de rug.
Hoe wordt het O'Donnell-Luria-Rodan syndroom behandeld?
Geen genezing
Er is geen behandeling die het O'Donnell-Luria-Rodan syndroom kan genezen. De behandeling is er op gericht om de ontwikkeling van kinderen met dit syndroom zo goed mogelijk te laten verlopen en bijkomende problemen zo vroeg mogelijk op te sporen en waar mogelijk te behandelen.
Blauwe lamp
Een deel van de baby's krijgt een behandeling voor geelzucht door middel van het liggen onder een blauwe lamp. Op deze manier kan het lichaam geholpen worden om de gele gekleurde afvalstoffen sneller uit het lichaam te laten verwijderen.
Kinderfysiotherapie
Een fysiotherapeut kan ouders tips en adviezen geven hoe ze hun kindje zo goed mogelijk kunnen stimuleren om er voor te zorgen dat de ontwikkeling zo optimaal als mogelijk verloopt.
Kinderlogopedie
Een logopediste kan tips en adviezen geven indien er problemen zijn met zuigen, drinken, kauwen of slikken. Ook kan de logopediste helpen om de spraakontwikkeling zo goed mogelijk te stimuleren. Praten kan ook ondersteund worden door middel van gebaren of pictogrammen. Op die manier kunnen kinderen zich leren uitdrukken ook als ze nog geen woorden kunnen gebruiken.

Kinderergotherapie
Een ergotherapeut kan tips en adviezen geven hoe de verzorging en de dagelijks activiteiten van een kind zo soepel mogelijk kunnen verlopen. Ook kan de ergotherapeut advies geven over materialen die de ontwikkeling van een kind kunnen stimuleren.
Revalidatiearts
Een revalidatiearts coördineert de verschillende therapieën en adviseert ook over hulpmiddelen zoals bijvoorbeeld een aangepaste buggy, een rolstoel, steunzolen of aangepaste schoenen.
Ook is het mogelijk via een revalidatie centrum naar een aangepaste peutergroep te gaan en daar ook therapie te krijgen en later op dezelfde manier onderwijs te gaan volgen.
School
De meeste kinderen met het O'Donnell-Luria-Rodan syndroom hebben extra begeleiding bij het leren nodig. Een deel van de kinderen kan regulier onderwijs volgen met behulp van ondersteuning. Een ander deel van de kinderen gaat uiteindelijk toch naar het speciaal onderwijs van cluster 2,3 of 4 omdat zij daar in kleinere klassen zitten en meer hulp en ook therapie kunnen krijgen.
Orthopedagoog
Een orthopedagoog kan ouders tips en adviezen geven hoe om gaan met problemen met bijvoorbeeld boos worden of het maken van contacten met andere kinderen.
Kinder- en jeugdpsychiater
Een kinder- en jeugdpsychiater kan advies geven hoe om te gaan met gedragsproblemen zoals ADHD, snel boos worden, ansgt of autisme. Soms is het nodig om gedrag regulerende medicatie zoals methylfenidaat voor ADHD of risperidonof aripiprazol voor prikkelovergevoeligheid te geven.
Aanvalsbehandeling epilepsie
De meeste epilepsieaanvallen gaan vanzelf over binnen enkele minuten. Omstanders hoeven dan niets te doen om de aanval te doen stoppen. Het is belangrijk om zo rustig mogelijk te blijven en het kind zo veel mogelijk met rust te laten.
Wanneer een aanval na 5 minuten nog niet vanzelf gestopt is, dan zal vaak geadviseerd worden om medicijnen te geven om een aanval te doen stoppen. De behandelende arts zal altijd aangeven welk tijdstip voor een bepaald kind het beste is. Medicijnen die gebruikt kunnen worden voor het stoppen van een aanval zijn diazepam rectiole (Stesolid®), midazolam neusspray, midazolam rectiole, lorazepam of clonazepam druppels.
Het effect van deze medicijnen ontstaat na enkele minuten. Nadien zal het kind meestal in slaap vallen, soms ook niet.

Behandeling epilepsie
Met behulp van medicijnen wordt geprobeerd om de epilepsieaanvallen zo veel mogelijk te voorkomen en het liefst er voor te zorgen dat er helemaal geen epilepsieaanvallen meer voorkomen. Meestal lukt het om de epilepsieaanvallen met een medicijn onder controle te krijgen, soms is een combinatie van medicijnen nodig.
Verschillende soorten medicijnen kunnen gebruikt worden om de epilepsie onder controle te krijgen. Er bestaat geen duidelijk voorkeursmedicijn. Medicijnen die vaak gebruikt worden zijn natriumvalproaat (Depakine ®), levetiracetam (Keppra ®), clobazam (Frisium ®) en zonisamide (Zonegran®).
Bij een deel van de kinderen zal het niet lukken om de epilepsieaanvallen met medicijnen onder controle te krijgen. Er bestaan ook andere behandelingen die een goed effect kunnen hebben op de epilepsie, zoals een ketogeen dieet, een nervus vagusstimulator, of een behandeling met methylprednisolon. Ook een combinatie van deze behandelingen met medicijnen die epilepsie onderdrukken is goed mogelijk.

Behandeling slaapproblemen
Een vast slaapritueel en een vast slaappatroon kunnen kinderen helpen om beter te kunnen slapen. Het medicijn melatonine kan helpen om beter in slaap te kunnen vallen. Er bestaan ook vormen van melatonine met vertraagde afgifte die ook kunnen helpen om weer in slaap te vallen wanneer kinderen in de nacht wakker worden. Slaapmiddelen worden liever niet gegeven aan kinderen omdat kinderen hier aangewend raken en niet meer zonder deze medicatie kunnen. Soms wordt het medicijn promethazine gebruikt om kinderen beter te kunnen laten slapen. Het is altijd belangrijk om uit te sluiten dat epilepsie de oorzaak is van de slaapproblemen, in geval van epilepsie is epilepsie behandeling nodig.

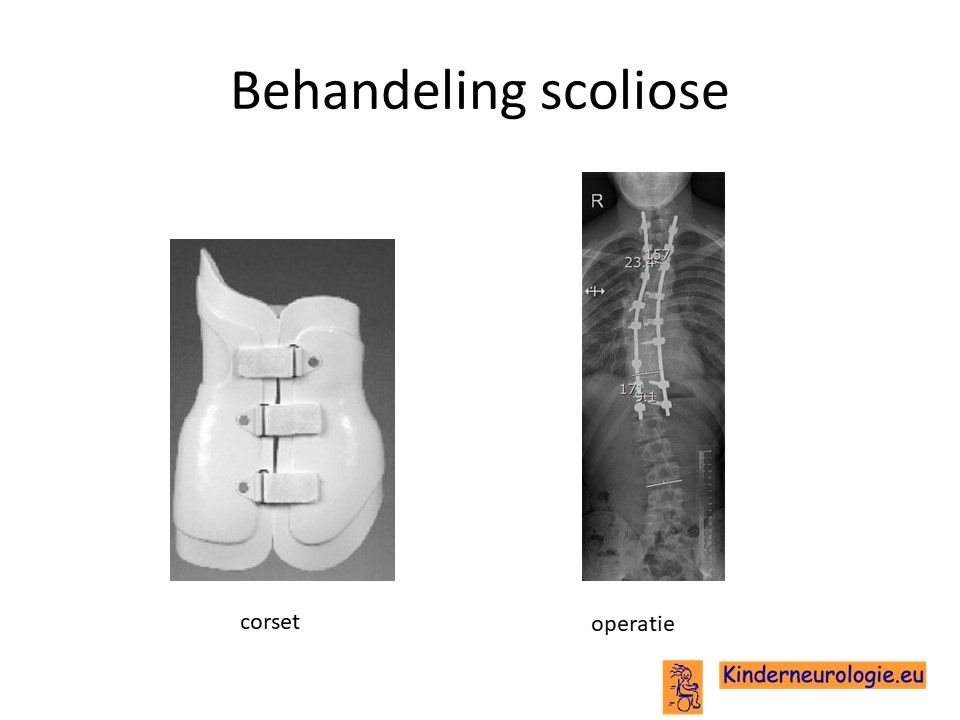
Scoliose
Lichte vormen van verkromming van de wervelkolom hebben geen behandeling nodig. Wanneer de verkromming toeneemt, kan deze behandeld worden met een gipscorset om verdergaande verkromming van de wervelkolom te voorkomen. Wanneer een gipscorset onvoldoende effect heeft, kan een operatie nodig zijn waarbij de wervels vastgezet. Deze behandeling wordt uitgevoerd door een orthopeed.
Kinderuroloog
Wanneer de zaadballen niet goed indalen, dan kan de kinderuroloog door middel van een operatie er voor zorgen dat de zaadballen wel in de balzak komen te liggen.
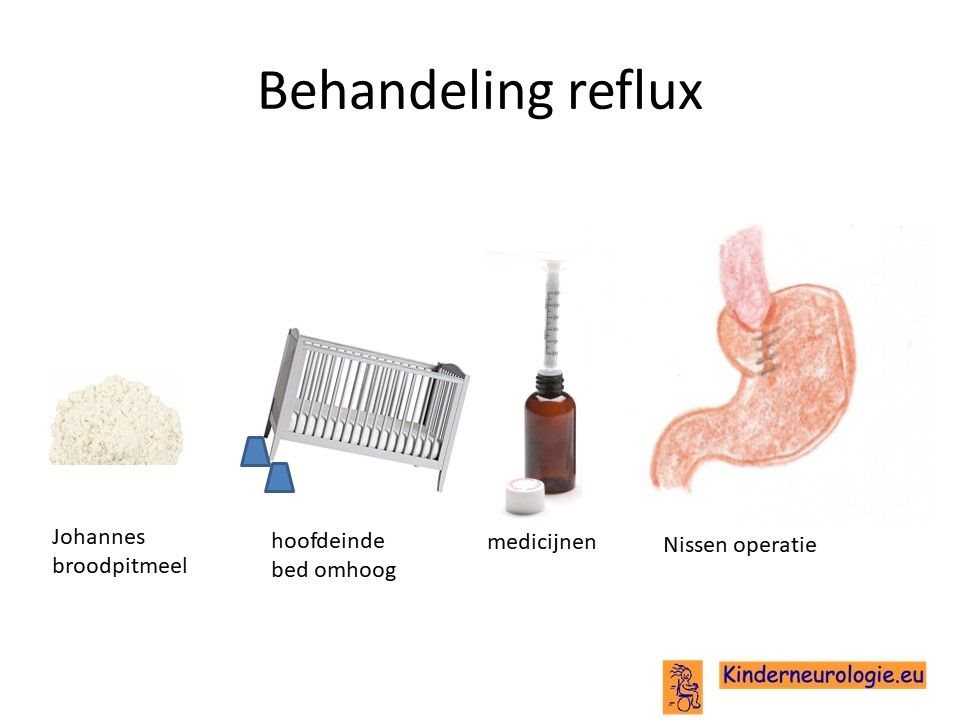
Reflux
Reflux kan er voor zorgen dat kinderen slecht eten. Door de voeding in te dikken met johannesbroodpitmeel kan de voeding minder gemakkelijk terug stromen van de maag naar de slokdarm. Ook zijn er medicijnen die de maaginhoud minder zuur kunnen maken waardoor de slokdarm minder geprikkeld wordt bij terugstromen van de maaginhoud. Medicijnen die hiervoor gebruikt worden zijn ranitidine, omeprazol of esomeprazol. Indien dit allemaal niet voldoende is, kan een operatie nodig zijn waarbij de overgang van de slokdarm naar de maag nauwer wordt gemaakt, waardoor de voeding ook minder gemakkelijk terug kan stromen. Dit wordt een Nissen-operatie genoemd.
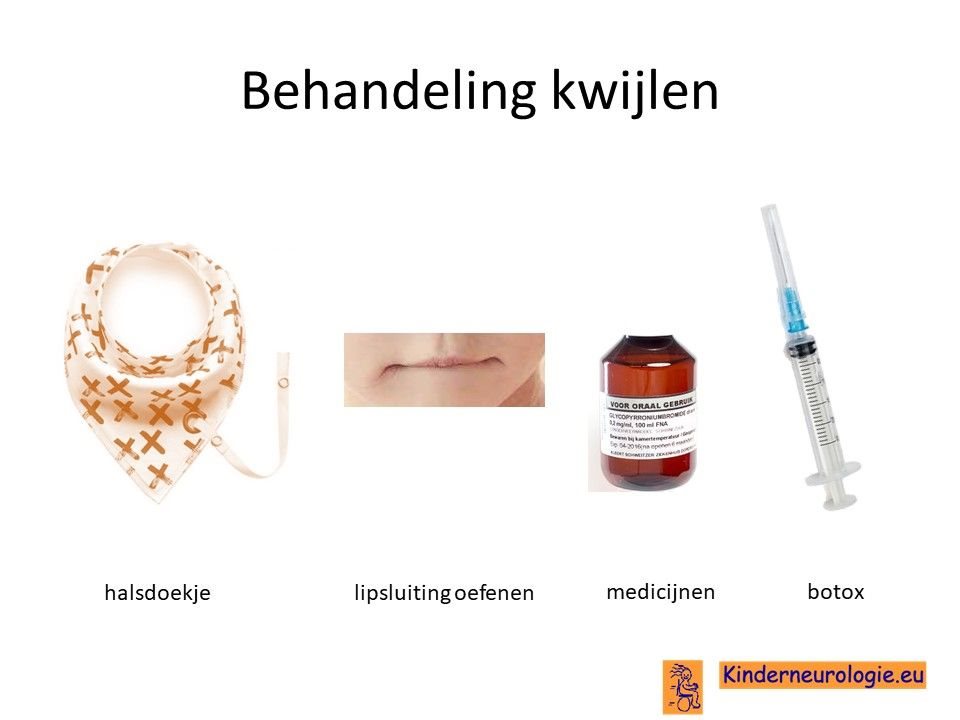
Kwijlen
Kwijlen kan verminderen door kinderen er bewust van te maken dat ze hun speeksel moeten doorslikken. Ook kunnen oefeningen waarbij geoefend wordt om de mond te sluiten helpen. Er bestaan moderne halsdoekjes die kwijl kunnen opvangen, zodat de kleding niet vies en nat wordt.
Er bestaan medicijnen die het kwijlen minder kunnen maken. Het meest gebruikte medicijn hierdoor is glycopyrrhonium. Soms kan een behandeling van de speekselklieren door middel van botox of door middel van een operatie nodig zijn om er voor zorgen dat kinderen minder kwijlen. Per kind zullen de voor- en nadelen van elke behandeling moeten worden afgewogen.
Verstopping van de darmen
Het medicijn macrogol kan er voor zorgen dat de ontlasting soepel en zacht blijft en stimuleert de darmwand om actief te blijven. Hierdoor kunnen kinderen gemakkelijker hun ontlasting kwijt. Verder blijft het belangrijk om te zorgen dat kinderen voldoende vocht en vezels binnen krijgen en zo veel als kan bewegen. Soms zijn zetpillen nodig om de ontlasting op gang te krijgen.

Financiële kant van zorg voor een kind met een beperking
De zorg voor een kind met een beperking brengt vaak extra kosten met zich mee. Er bestaan verschillende wetten die zorg voor kinderen met een beperking vergoeden.
Daarnaast bestaan regelingen waar ouders een beroep op kunnen doen, om een tegemoetkoming te krijgen voor deze extra kosten. Meer informatie hierover vindt u in de folder financiën kind met een beperking.

Begeleiding
Een maatschappelijk werkende of psycholoog kan begeleiding geven hoe het hebben van deze aandoening een plaats kan krijgen in het dagelijks leven. Het kost vaak tijd voor ouders om te verwerken dat de toekomstverwachtingen van hun kind er anders uit zien dan waarschijnlijk verwacht was. Ook vinden veel ouders het vaak lastig hoe zij hun tijd en aandacht moeten verdelen tussen het kind met de beperking en andere kinderen in het gezin. In de folder aandacht en tijd voor brussen vindt u tips die u hierbij kunnen helpen.

Door middel van een oproepje op het forum van deze site kunt u proberen in contact te komen met andere kinderen en hun ouders/verzorgers die ook te maken hebben met het O'Donnell-Luria-Rodan syndroom.
Wat is de prognose van het O'Donnell-Luria-Rodan syndroom?
Blijvende problemen
Kinderen die een ontwikkelingsachterstand hebben als gevolg van het O'Donnell-Luria-Rodan syndroom, blijven deze problemen vaak houden op volwassen leeftijd. Een deel van de volwassenen kan zelfstandig functioneren. Een ander deel van de volwassenen heeft in meer of mindere mate de hulp van anderen nodig hebben om te kunnen functioneren in het dagelijks leven.
Transitie van zorg
Tussen de leeftijd van 16 en 18 jaar wordt de zorg vaak overgedragen van kinderspecialisten naar specialisten die de zorg aan volwassenen geven. Het is belangrijk om tijdig hierover na te denken. Is er behoefte de zorg over te dragen naar specialisten voor volwassenen of kan de huisarts de zorg leveren die nodig is. En als er behoefte is aan overdragen van de zorg naar specialisten voor volwassenen, naar welke dokter(s) wordt de zorg dan overgedragen? In welk ziekenhuis kan de zorg het beste geleverd worden. Het proces van overdragen van de zorg wordt transitie genoemd. Het is belangrijk hier tijdig over na te denken en een plan voor te maken samen met de dokters die betrokken zijn bij de zorg op de kinderleeftijd. Ook verandert er veel in de zorg wanneer een jongere de leeftijd van 18 jaar bereikt. Voor meer informatie over deze veranderingen verwijzing wij u naar het artikel veranderingen in de zorg 18+
![]()
Volwassenen
Omdat deze ziekte nog niet heel lang bekend is, is er niet heel veel bekend over volwassenen met deze aandoening.
Levensverwachting
De levensverwachting van kinderen en volwassenen met het O'Donnell-Luria-Rodan syndroom hangt sterk samen met de klachten die worden veroorzaakt door het O'Donnell-Luria-Rodan syndroom. Voor de meeste kinderen zal de levensverwachting niet anders zijn dan van kinderen zonder dit syndroom. De levensverwachting kan worden verkort als gevolg van een moeilijk behandelbare vorm van epilepsie.
Kinderen
Kinderen van een volwassene met het O'Donnell-Luria-Rodan syndroom zelf 50% kans om zelf ook het O'Donnell-Luria-Rodan syndroom te krijgen. Vanwege hun verstandelijke beperking zal een groot deel van de volwassenen met dit syndroom niet zelf kinderen gaan krijgen. Of eventuele kinderen evenveel, meer of minder klachten zullen hebben als de ouder valt van te voren niet te voorspellen. Indien de volwassene geen kinderen wil of kan krijgen, moet wellicht nagedacht moeten worden over anticonceptie, waarover u in deze folder meer informatie vindt.
Hebben broertjes en zusjes ook een verhoogde kans om ook het O'Donnell-Luria-Rodansyndroom te krijgen?
Het O'Donnell-Luria-Rodan syndroom wordt veroorzaakt door een fout in het erfelijke materiaal van het 7e chromosoom. Vaak is deze fout bij het kind zelf ontstaan en niet overgeërfd van een van de ouders. Broertjes en zusjes hebben daarom een nauwelijks verhoogde kans om zelf ook het O'Donnell-Luria-Rodan syndroom te krijgen. Dit zou alleen kunnen indien een van de ouders de fout in een eicel of zaadcel heeft zitten zonder dat dit foutje ergens anders in de lichaamscellen voorkomt. Dit wordt ouderlijke mocaïsisme genoemd. De kans hierop is heel klein, ongeveer 1%.
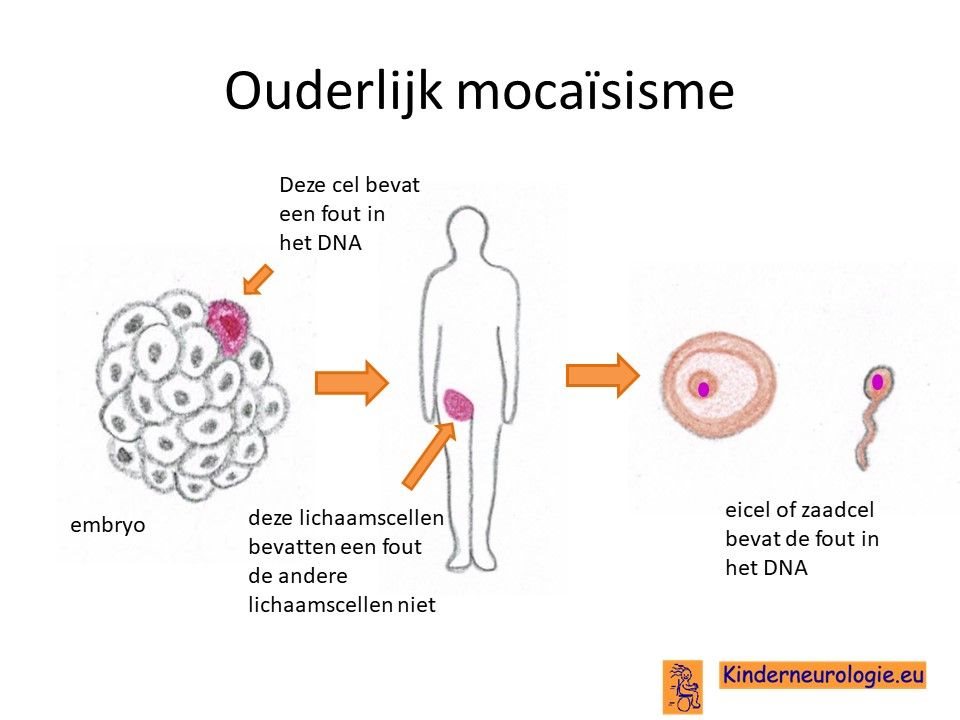
Wanneer een van de ouders zelf het O'Donnell-Luria-Rodan syndroom heeft, dan hebben broertjes en zusjes 50% kans om ook zelf dit syndroom te krijgen.
Een klinisch geneticus kan hier meer informatie over geven.
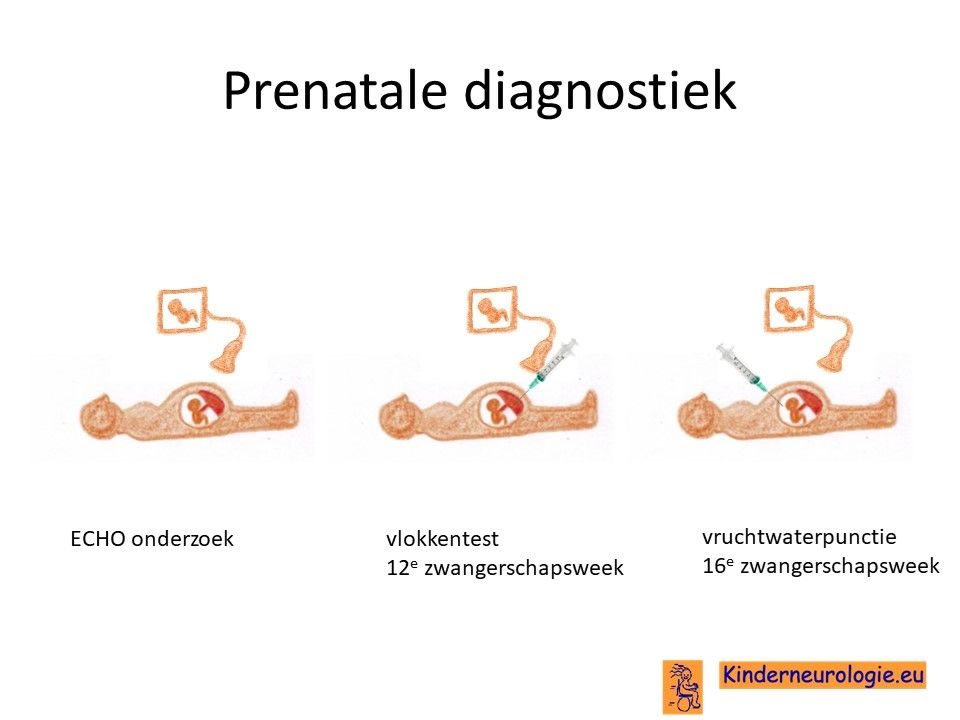
Prenatale diagnostiek
Wanneer bekend is welk foutje in een familie heeft gezorgd voor het ontstaan van het O'Donnell-Luria-Rodan syndroom, dan is het mogelijk om tijdens een zwangerschap prenatale diagnostiek te verrichten in de vorm van een vlokkentest of een vruchtwaterpunctie om te kijken of dit kindje ook het O'Donnell-Luria-Rodansyndroom heeft. Beide ingrepen hebben een klein risico op het ontstaan van een miskraam (0,5% bij de vlokkentest en 0,3% bij de vruchtwaterpunctie).
Wilt u ook uw verhaal kwijt, dat kan: verhalen kunnen gemaild worden via via contact en zullen daarna zo spoedig mogelijk op de site worden geplaatst. Voor meer informatie zie hier.
Heeft u foto's die bepaalde kenmerken van deze aandoening duidelijk maken en die hier op de website mogen worden geplaatst, dan vernemen wij dit graag via contact.
Referenties
- Heterozygous Variants in KMT2E Cause a Spectrum of Neurodevelopmental Disorders and Epilepsy. O'Donnell-Luria AH, Pais LS, Faundes V, Wood JC, Sveden A, Luria V, Abou Jamra R, Accogli A, Amburgey K, Anderlid BM, Azzarello-Burri S, Basinger AA, Bianchini C, Bird LM, Buchert R, Carre W, Ceulemans S, Charles P, Cox H, Culliton L, Currò A; Deciphering Developmental Disorders (DDD) Study, Demurger F, Dowling JJ, Duban-Bedu B, Dubourg C, Eiset SE, Escobar LF, Ferrarini A, Haack TB, Hashim M, Heide S, Helbig KL, Helbig I, Heredia R, Héron D, Isidor B, Jonasson AR, Joset P, Keren B, Kok F, Kroes HY, Lavillaureix A, Lu X, Maas SM, Maegawa GHB, Marcelis CLM, Mark PR, Masruha MR, McLaughlin HM, McWalter K, Melchinger EU, Mercimek-Andrews S, Nava C, Pendziwiat M, Person R, Ramelli GP, Ramos LLP, Rauch A, Reavey C, Renieri A, Rieß A, Sanchez-Valle A, Sattar S, Saunders C, Schwarz N, Smol T, Srour M, Steindl K, Syrbe S, Taylor JC, Telegrafi A, Thiffault I, Trauner DA, van der Linden H Jr, van Koningsbruggen S, Villard L, Vogel I, Vogt J, Weber YG, Wentzensen IM, Widjaja E, Zak J, Baxter S, Banka S, Rodan LH. Am J Hum Genet. 2019;104:1210-1222.
Links
www.zeldsamen.nl
(netwerken voor zeldzame genetische syndromen)
Laatst bijgewerkt: 25 november 2020
Auteur: JH Schieving