Wat is het CAMTA1-syndroom?
Het CAMTA1 syndroom is een aangeboren aandoening waarbij kinderen problemen hebben met hun evenwicht al dan niet in combinatie met een ontwikkelingsachterstand.
Hoe wordt het CAMTA1-syndroom ook wel genoemd?
CAMTA1 is de naam van de plaats op het erfelijk materiaal waar het foutje gevonden is. Er wordt ook gesproken van het CAMTA1-gerelateerd syndroom. Gerelateerd betekent verband houdend met.
Niet progressieve ataxie
Het CAMTA1 syndroom is een vorm van een groep aandoeningen die ook wel niet-progressieve ataxieën worden genoemd. Ataxie is een andere naam voor een evenwichtsstoornis. Met niet-progressief wordt bedoeld dat kinderen deze evenwichtsstoornis hebben en dat deze evenwichtsstoornis niet verergerd gedurende het leven. De Engelse benaming hiervoor is non-progressieve cerebellair ataxia, ook wel afgekort met de letters NPCA.
CANPMR
Een nieuwe naam voor CAMTA1-syndroom is CANPMR. De letters CA staan voor cerebellaire ataxie, de medische naam voor de evenwichtsproblemen die kinderen met dit syndroom hebben. De N staat voor non-progressief en geeft aan dat deze problemen niet toenemen in de loop van de tijd. De letters PMR staan voor psychomotore retardatie, het medische woord voor een ontwikkelingsachterstand.
CECBA
Een andere naam die tegenwoordig ook gebruikt wordt is CECBA. Dit staat voor Cerebellaire dysfunctie met variabele cognitieve en gedragsmatige problemen (behavioral abnormalities).
Hoe vaak komt CAMTA1-syndroom voor bij kinderen?
Het is niet goed bekend hoe vaak het CAMTA1 syndroom bij kinderen voorkomt. Het syndroom is nog maar sins 2012 bekend. Daarom zal nog niet bij alle kinderen met dit syndroom ontdekt zijn dat ze dit syndroom hebben. Geschat wordt dat deze aandoening bij minder dan één op de 100.000 kinderen voorkomt.

Bij wie komt het CAMTA1-syndroom voor?
Het CAMTA1-syndroom is al voor de geboorte aanwezig. Meestal wordt tijdens het eerste of het tweede levensjaar duidelijk dat de ontwikkeling van kinderen met dit syndroom anders verloopt dan van andere kinderen zonder dit syndroom.
Zowel jongens als meisjes kunnen dit syndroom krijgen.
Wat is de oorzaak van het CAMTA1-syndroom?
Fout in het erfelijk materiaal
Het CAMTA1-syndroom wordt veroorzaakt door een fout in het erfelijk materiaal van chromosoom 1. De plaats van deze fout wordt het CAMTA1-gen genoemd.
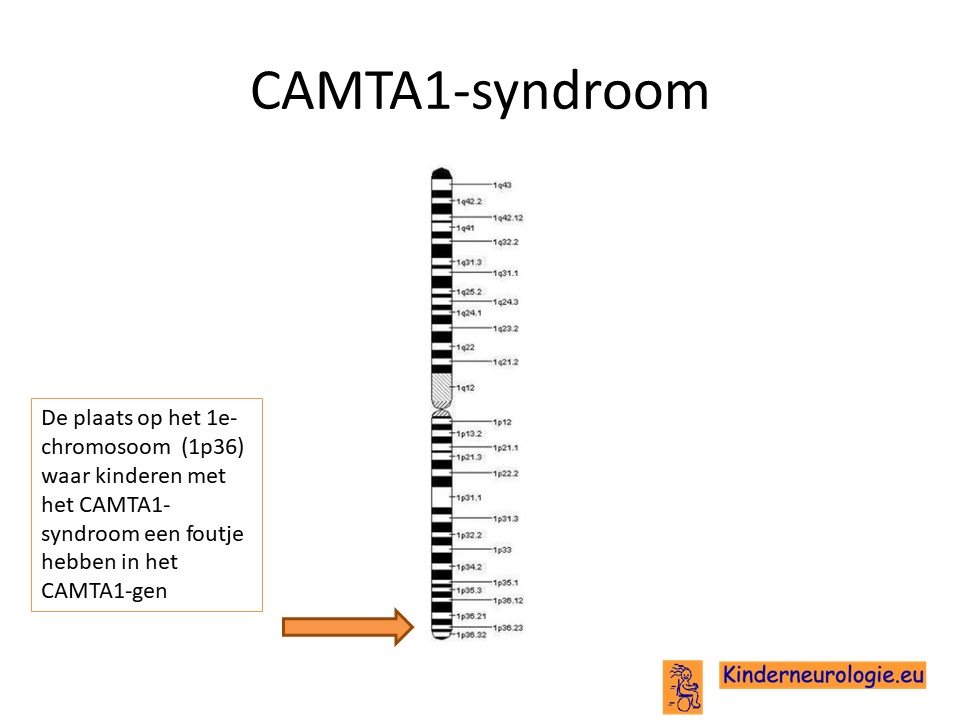
Autosomaal dominant
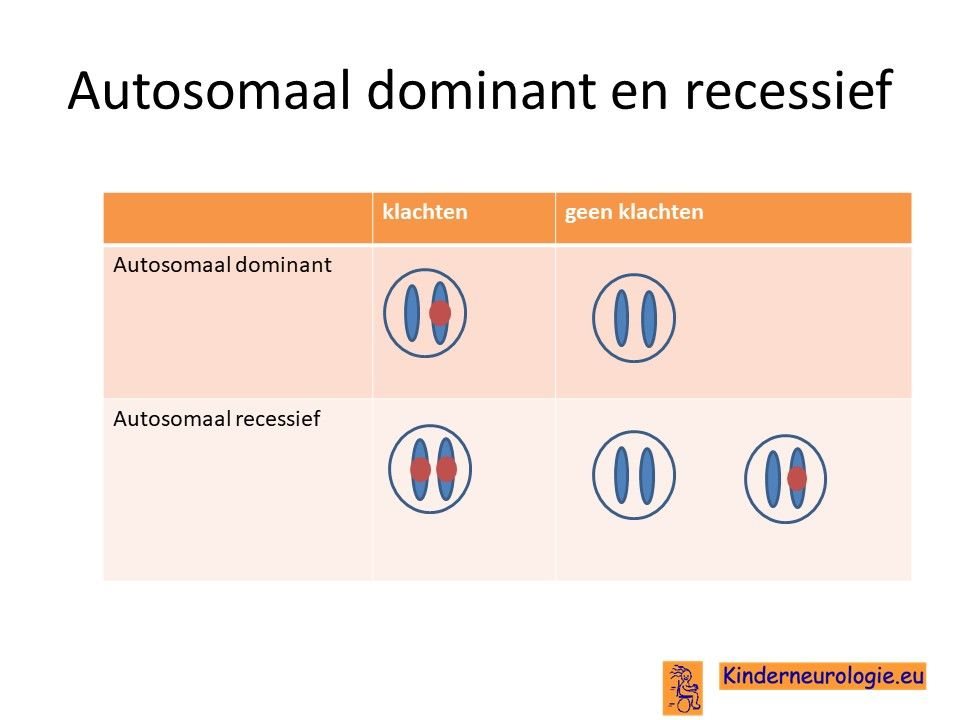
Het CMATA1-syndroom is een autosomaal dominante aandoening. Dit houdt in dat een fout op een chromosoom 1 op de plaats van het CAMTA1- gen al voldoende is om de aandoening te krijgen. Dit in tegenstelling tot een autosomaal recessieve aandoening waarbij twee fouten op beide chromosomen 1 nodig zijn om de ziekte te krijgen.
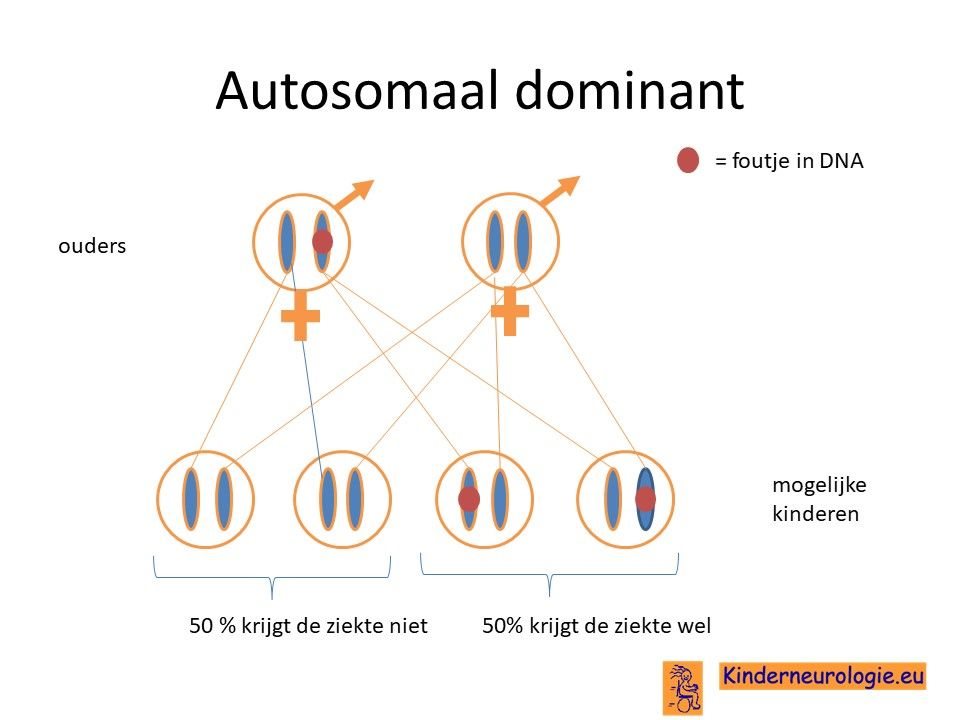
Overgeërfd van een ouder
Een deel van de kinderen heeft de aandoening geërfd van de vader of de moeder die zelf ook het CAMTA1- syndroom heeft. Soms is al wel bekend dat de vader of moeder ook problemen heeft, soms ook niet omdat sommige mensen maar weinig problemen hebben als gevolg van dit syndroom.
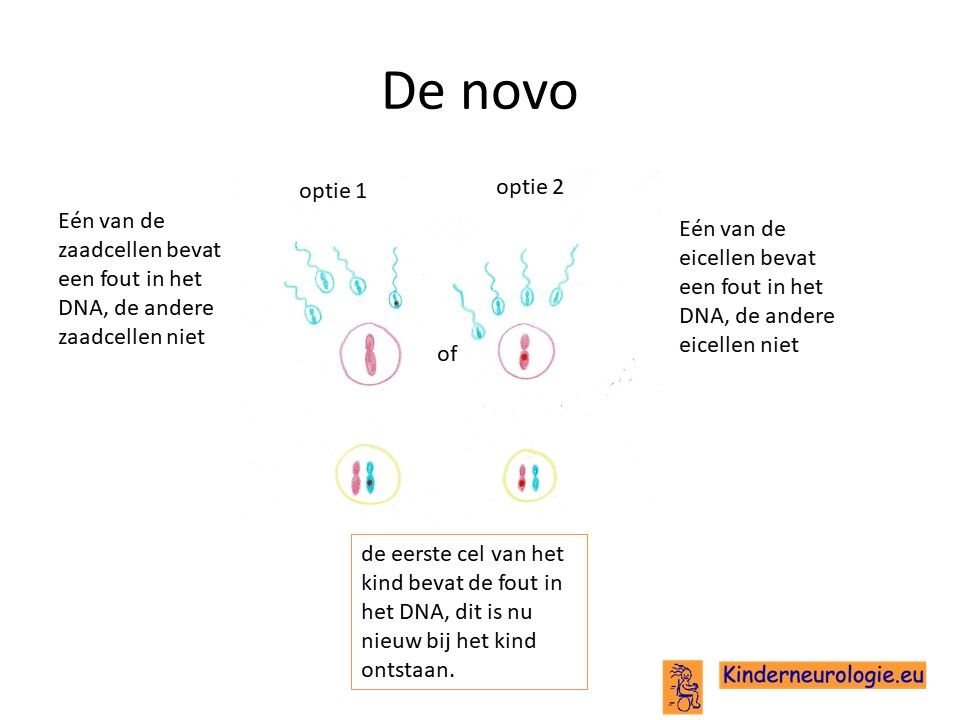
Bij het kind zelf ontstaan
Het kan ook zijn dat het foutje in het CAMTA1-gen op chromosoom 1 bij het kind zelf ontstaan is na de bevruchting van de eicel door de zaadcel. Dit wordt ook wel de novo, nieuw ontstaan genoemd.
Omdat deze aandoening nog maar kort bekend is, is niet bekend hoe vaak deze fout bij het kind zelf ontstaan is en hoe vaak de fout overgeërfd is van een ouder.
Type fout in het DNA
Het type fout in het DNA bepaalt ook hoeveel en welke klachten een kind zal gaan krijgen. Bepaalde fouten zorgen er voor dat er helemaal geen werkzaam eiwit meer wordt gemaakt, deze fouten hebben de grootste gevolgen voor een kind. Bij ander type fouten wordt er een afwijkend maar nog wel werkzaam eiwit gemaakt of is er minder goed werkzaam eiwit dan gebruikelijk. Deze kinderen zullen minder klachten hebben dan kinderen die helemaal geen werkend eiwit meer hebben.

Afwijkend eiwit
Door het foutje in het erfelijk materiaal wordt een bepaald eiwit niet goed aangemaakt. Dit eiwit wordt het CAMTA1-eiwit genoemd. CAMTA1 staat voor calmoduline binding transcription activator -1. Dit eiwit speelt een belangrijke rol bij de aanleg van de kleine hersenen. Bij kinderen met het CAMTA1-syndroom worden met name de kleine hersenen anders aangelegd dan gebruikelijk.

Wat zijn de symptomen van het CAMTA1-syndroom?
Variatie
Er bestaat een grote variatie in hoeveelheid en ernst van onderstaande symptomen. Sommige kinderen hebben een paar symptomen, anderen hebben er meer. Het valt van te voren niet te voorspellen van hoeveel symptomen een kind last zal gaan krijgen.

Jouw kind is uniek
Bedenk dat onderstaande symptomen kunnen voorkomen bij jouw kind, maar ook niet allemaal zullen voorkomen. Jouw kind is uniek en veel meer dan een kind met deze aandoening. Het lezen van mogelijke symptomen die kunnen voorkomen, kan ouders het gevoel geven dat er alleen maar aandacht is voor de beperkingen van het kind. Dat is zeer zeker niet de bedoeling. Jouw kind is bijvoorbeeld lief, grappig, gevoelig, gezellig,sociaal, vindingrijk, nieuwsgierig, ondeugend, enthousiast,een zonnestraaltje, creatief en/of innemend en dat vind je niet terug in onderstaande symptomen die kunnen horen bij dit syndroom. Dat kan ook niet, want die eigenschappen maken jouw kind nu eenmaal uniek. Blijf daar vooral naar kijken en zie deze symptomen meer als achtergrondinformatie die je kunnen helpen om te begrijpen wat er met je kind aan de hand zou kunnen zijn wanneer jouw kind zich anders ontwikkelt of ergens last van heeft. Deze informatie kan jullie als ouders en hulpverleners een handvat geven wat hiervoor een mogelijke verklaring kan zijn.

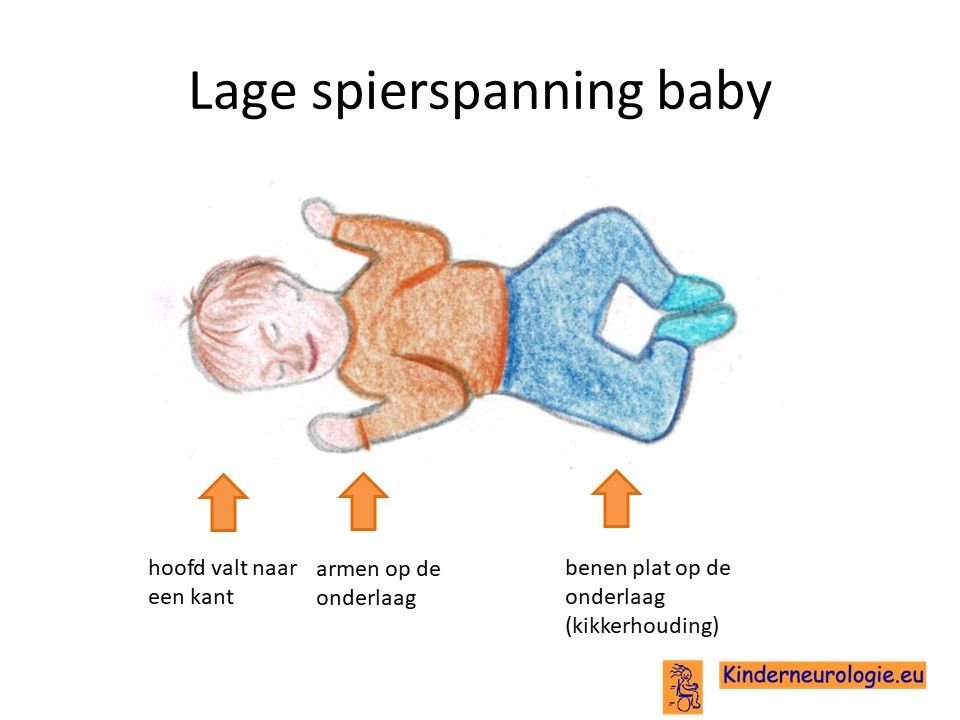
Lage spierspanning
Kinderen met het CAMTA1-syndroom hebben vaak een lage spierspanning wanneer ze geboren worden. Ze voelen slapper aan dan andere kinderen van hun leeftijd en moeten goed ondersteund worden, wanneer ze opgetild worden. Omdat ook de nekspieren slaper zijn, hebben veel kinderen moeite om hun hoofd op te tillen. Een te lage spierspanning wordt ook wel hypotonie genoemd.
Tragere ontwikkeling
Kinderen met het CAMTA1-syndroom ontwikkelen zich vaak trager dan hun leeftijdsgenoten. Ze gaan later rollen, zitten, kruipen, staan en lopen dan hun leeftijdsgenoten. De meeste kinderen leren los lopen tussen de leeftijd van 1,5 en 2 jaar.
Ook de taalontwikkeling komt vaak later op gang. De eerste woordjes komen vaak pas laat rond de leeftijd van 3 jaar.
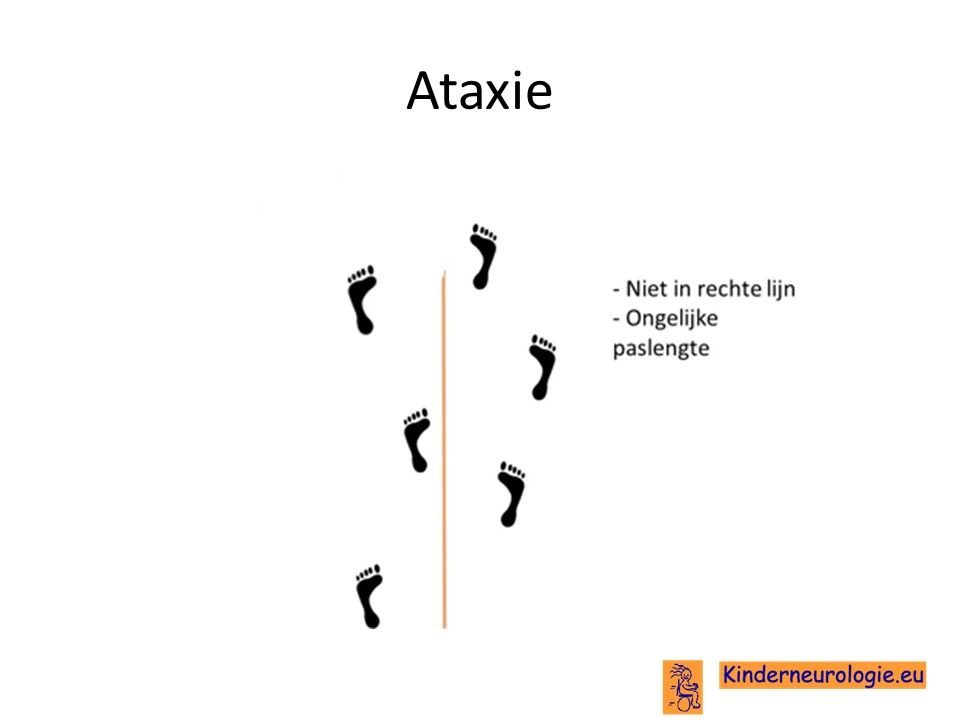
Ataxie
Kinderen met het CAMTA1-syndroom hebben problemen met het bewaren van hun evenwicht. Dit evenwichtsprobleem wordt ataxie genoemd. Kinderen vallen gemakkelijker dan andere kinderen. Vaak zetten kinderen hun voeten verder uit elkaar om zo steviger te kunnen staan en te lopen. Dit wordt ook wel een breedbasisch looppatroon genoemd. Kinderen zetten de ene keer een te grote pas en de andere keer juist een kleine pas. De meeste kinderen zijn ondanks de problemen met het bewaren van het evenwicht goed in staat om zonder hulp te kunnen lopen.
Door de ataxie hebben kinderen ook moeite met het pakken van een voorwerp. Vaak grijpen ze naast het voorwerp. Dit wordt een dysmetrie genoemd. Vaak kost het meer tijd om het voorwerp uiteindelijk te pakken te krijgen.
Tremor
De handen kunnen een trillende beweging maken wanneer kinderen wat willen pakken. Daardoor wordt het bijvoorbeeld moeilijker om te schrijven, een kopje naar de mond te brengen of knoopjes dicht te maken. Deze trillende beweging wordt tremor genoemd.

Spasticiteit
Met het ouder worden krijgt een klein deel van de kinderen een verhoogde spierspanning, met name in de benen. Een te hoge spierspanning wordt spasticiteit genoemd. Spasticiteit in de benen kan er voor zorgen dat kinderen op hun tenen gaan lopen, waardoor het moeilijker is om het evenwicht te bewaren. Ook krijgen deze benen de neiging voor elkaar langs te kruizen wat het lopen moeilijker kan maken. Aanvallen met plostelinge toename van spierspanning kunnen voorkomen, dit wordt een spasme genoemd. Spasmes komen vooral voor bij vermoeidheid, ziek zijn, schrik of stress.

Problemen met praten
Een deel van de kinderen met het CAMTA1-syndroom praat minder duidelijk dan leeftijdsgenoten. Dit wordt dysartrie genoemd.
Problemen met leren
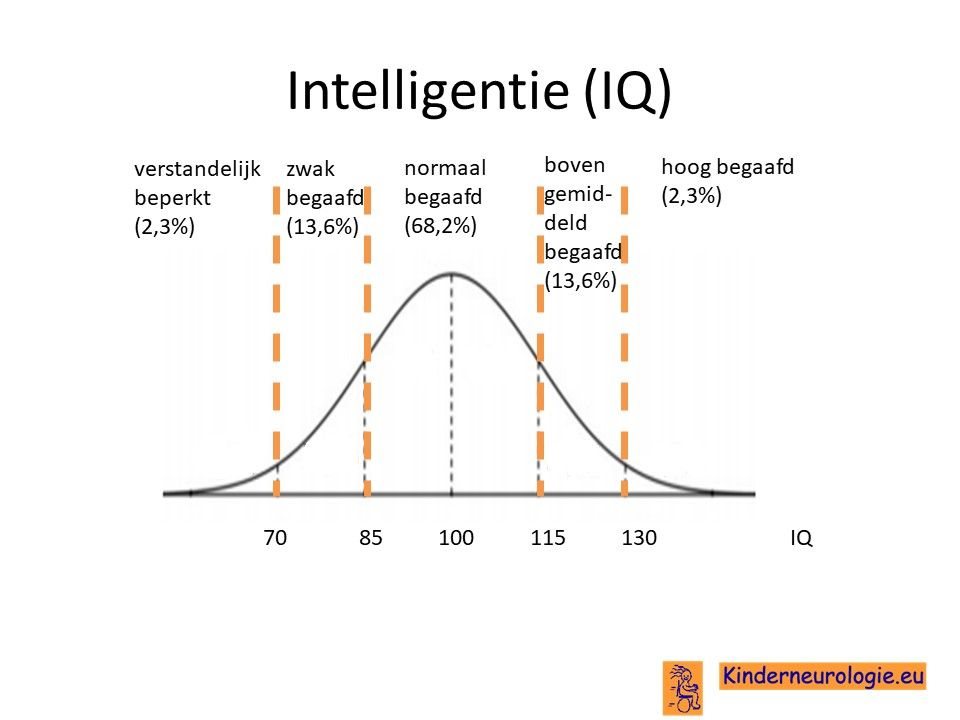
Een deel van de kinderen met het CAMTA1-syndroom heeft een normale intelligentie. Een ander deel van de kinderen met het CAMTA1-syndroom heeft een verminderde intelligentie (met een IQ-score tussen de 50 en 70). Vaak ligt het werktempo van kinderen met het CAMTA1-syndroom lager dan bij kinderen zonder dit syndroom. Een deel van de kinderen heeft moeite met ruimtelijk inzicht en met abstract denken. Ook onthouden is vaak lastiger voor kinderen met dit syndroom. Opvallend is vaak dat kinderen veel beter woorden kunnen onthouden, dan beelden en plaatjes.
Aandacht- en concentratiestoornissen
Kinderen met het CAMTA1-syndroom hebben vaker problemen met het vasthouden van de aandacht en concentratie. Ze zijn sneller afgeleid. Een deel van de kinderen krijgt ook de diagnose AD(H)D.
Autistiforme kenmerken
Een deel van de kinderen heeft autistiforme kenmerken. Deze kinderen houden van vaste structuur en voorspelbaarheid en kunnen moeilijk om gaan met veranderingen daarin. Soms kunnen kinderen daardoor heel boos worden. Ook vinden ze het vaak moeilijker om vriendjes en vriendinnetjes te maken. De kinderen zijn wat meer op zichzelf en daar tevreden mee.

Epilepsie
Een klein deel van de kinderen met het CAMTA1-syndroom heeft last van epilepsieaanvallen. Verschillende soorten epilepsieaanvallen kunnen voorkomen, zoals aanvallen met staren, aanvallen met verstijven van een arm of been (tonische aanvallen) of aanvallen met schokken van een arm of been (clonische aanvallen).
Uiterlijke kenmerken
Bij veel syndromen hebben kinderen vaak wat veranderde uiterlijke kenmerken. Hier hebben kinderen zelf geen last van, maar het kan de dokters helpen om te herkennen dat er sprake is van een syndroom en mogelijk ook van welk syndroom. Ook maakt dit vaak dat kinderen met hetzelfde syndroom vaak meer op elkaar lijken dan op hun eigen broertjes en zusjes, terwijl de kinderen toch niet familie van elkaar zijn.
Kinderen met het CAMTA1-syndroom hebben vaak een langwerpige vorm van hun gezicht. Het voorhoofd is hoog en breed. De ogen kunnen wat verder uit elkaar staan dan gebruikelijk. Ook lopen de ogen vaak wat omlaag in de richting van de oren. De neus is vaak plat met een stevige neuspunt en naar buiten gedraaide neusvleugels. De afstand tussen de neus en de mond kan vergroote zijn. De mond is vaak smal. De bovenlip is vaak dunner dan de onderlip. Er kan ruimte aanwezig zijn tussen de tanden. De kin is vaak smal en steekt een stukje naar voren toe. De oren staan vaak wat lager op het hoofd dan gebruikelijk.
Kleinere hoofdomtrek
Een deel van de kinderen met het CAMTA1-syndroom heeft een kleinere hoofdomtrek dan gebruikelijk.
Problemen met zien
Scheelzien komt vaak voor bij kinderen met deze aandoening. Bij een deel van de kinderen maken de ogen schokkende bewegingen. Dit wordt nystagmus genoemd. Kinderen hebben hier zelf geen last van, de hersenen zorgen dat kinderen gewoon een stilstand beeld krijgen te zien.
Problemen met slikken
Een deel van de kinderen heeft problemen slikken. Kinderen met het CAMTA1-syndroom verslikken zich gemakkelijker.
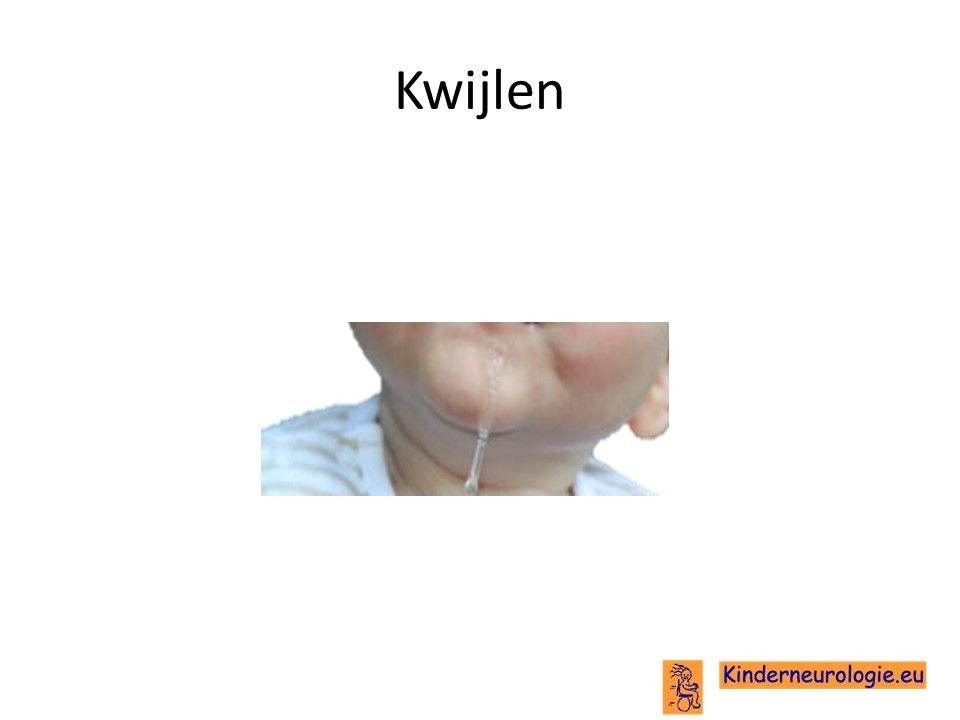
Kwijlen
Kinderen met dit syndroom hebben gemakkelijk last van kwijlen. Dit komt door slapheid van de spieren in het gezicht en rondom de mond, waardoor het speeksel gemakkelijk uit de mond loopt.
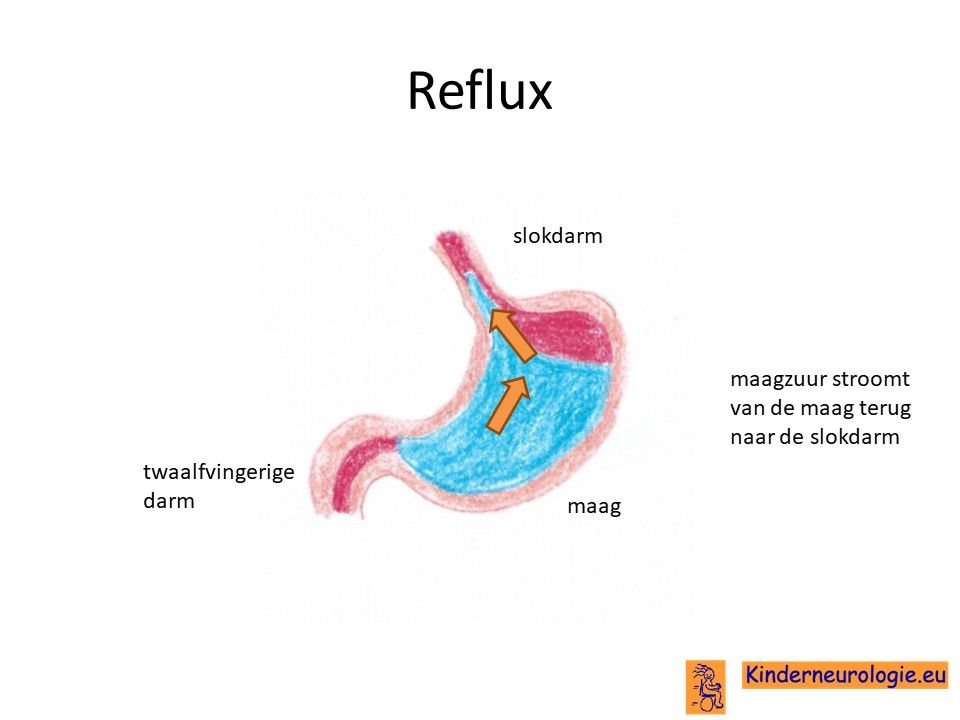
Reflux
Kinderen met dit syndroom hebben vaak last van het terugstromen van voeding vanuit de maag naar de slokdarm. Dit wordt reflux genoemd. Omdat de maaginhoud zuur is, komt het zuur zo ook in de slokdarm, soms zelfs ook in de mond. Dit zuur kan zorgen voor pijnklachten, waardoor kinderen moeten huilen en soms ook niet willen eten. Ook kan het maken dat kinderen moeten spugen.
Door het zuur kan de slokdarm geïrriteerd en ontstoken raken. Wanneer dit niet tijdige ontdekt en behandeld wordt, kan dit zorgen voor het spuug met daarin bloedsliertjes.
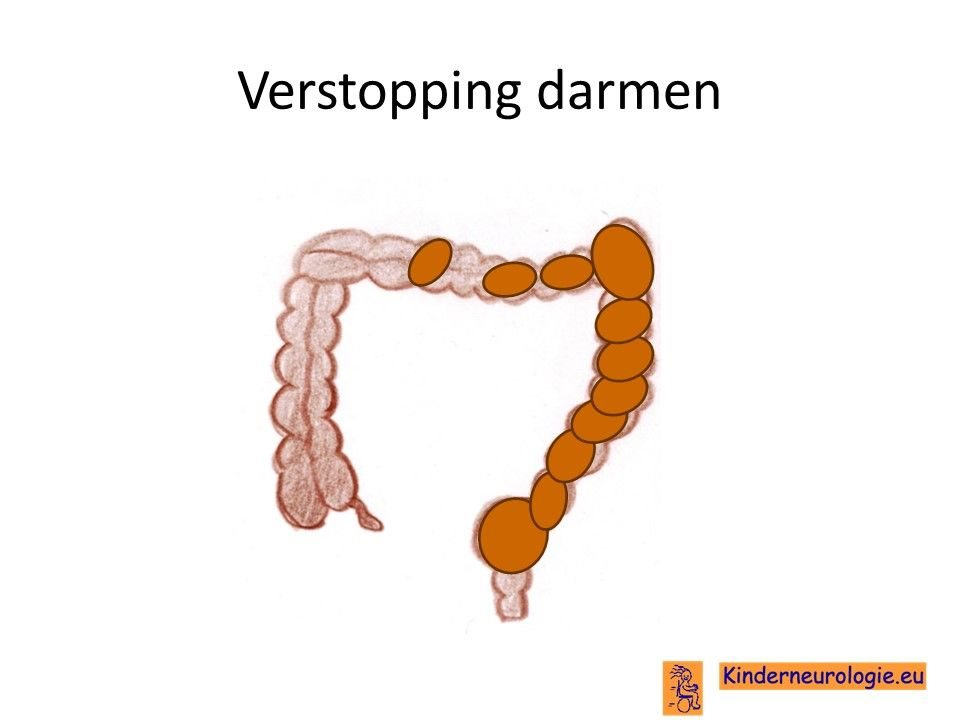
Verstopping van de darmen
Verstopping van de darmen komt vaak voor bij kinderen met dit syndroom. De ontlasting komt dan niet elke dag en is vaak hard waardoor kinderen moeite hebben met poepen. Dit kan buikpijnklachten geven.
Zindelijkheid
Het duurt vaak langer dan gebruikelijk voordat kinderen met dit syndroom zindelijk worden. De meeste kinderen worden dit uiteindelijk wel uit zichzelf.

Hoe wordt de diagnose CAMTA1-syndroom gesteld?
Verhaal en onderzoek
Op grond van het verhaal van een kind wat slapper is dan leeftijdsgenoten, zich trager ontwikkeld en problemen met het evenwicht heeft, kan worden vermoed dat de kleine hersenen anders werken dan gebruikelijk. Dit kan veel verschillende oorzaken hebben. Er zal ander aanvullend onderzoek nodig zijn om de juiste diagnose te stellen.
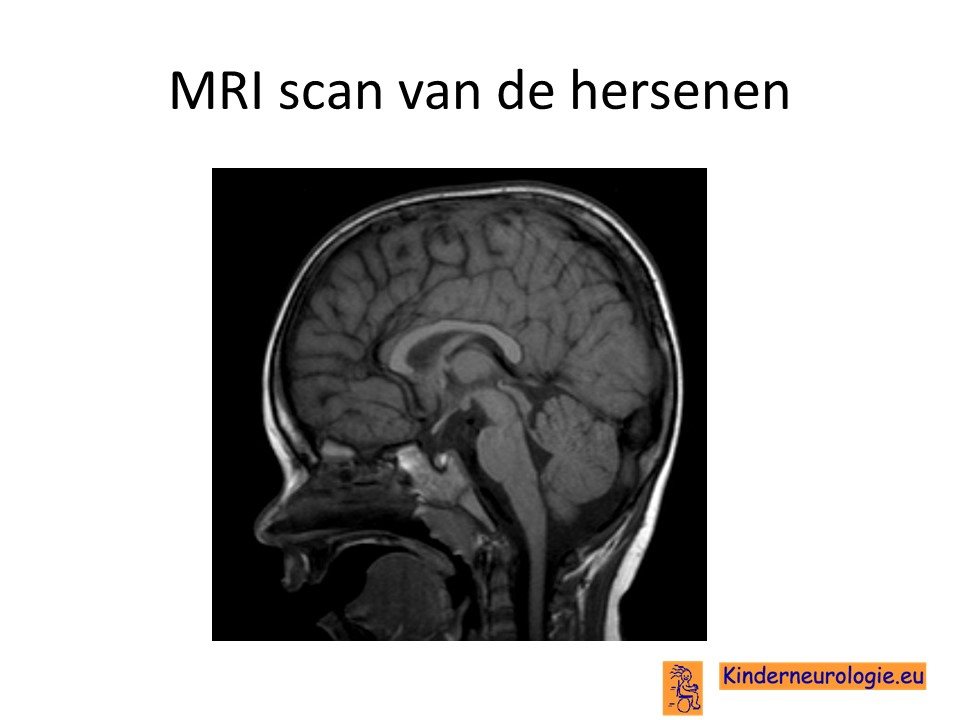
MRI van de hersenen
Wanneer er gedacht wordt aan een stoornis in het functioneren van de kleine hersenen, dan zal vaak een MRI scan van de hersenen gemaakt worden. Bij een deel van de kinderen met het CAMTA1-syndroom worden geen afwijkingen gezien op deze MRI-scan. Bij een ander deel van de kinderen is te zien dat de kleine hersenen anders zijn aangelegd dan gebruikelijk. Ook kunnen andere delen van de hersenen minder ontwikkeld zijn dan gebruikelijk zoals de hippocampus die een belangrijke rol bij onthouden speelt en de zogenaamde parietaal kwab die een belangrijke rol bij het registreren van gevoel en het ruimtelijk inzicht speelt.
Onderzoek erfelijk materiaal
Met behulp van een Array onderzoek kunnen alle chromosomen tegelijkertijd worden onderzocht om te kijken of er stukjes chromosoom missen of te vel aanwezig zijn. Op deze manier lukt het bij een deel van de kinderen om het CAMTA1-syndroom op te sporen. Wanneer aan dit syndroom gedacht wordt, kan er ook voor gekozen worden om met gericht DNA-onderzoek naar dit gen te kijken. Tegenwoordig wordt de diagnose CAMTA1-syndroom vaak gesteld met behulp van een nieuw genetisch onderzoek (exome sequencing) waarbij in een keer naar heel veel mogelijke fouten in het DNA gekeken kan worden. De diagnose kan dan gesteld worden zonder dat er specifiek naar gezocht is.
EEG
Kinderen met epilepsie krijgen vaak een EEG om te kijken van welk soort epilepsie er sprake is. Op het EEG worden vaak epileptiforme afwijkingen gezien. Deze afwijkingen zijn niet kenmerkend voor het CAMTA1-syndroom, maar kunnen bij veel andere syndromen met epilepsie ook gezien worden.
Hoe wordt het CAMTA1-syndroom behandeld?
Geen genezing
Er bestaat geen behandeling die het CAMTA1-syndroom kan genezen. De behandeling is er op gericht om zo goed mogelijk om te gaan met de gevolgen van het hebben van dit syndroom.
Kinderarts
In Nederland wordt door de zorg voor kinderen met een zeldzaam syndroom vaak gecoordineerd door de kinderarts in de eigen woonomgeving. Daarnaast kunnen kinderen ook begeleid worden door een speciale kinderarts die zich gespecialiseerd heeft in de zorg voor kinderen met een aangeboren en vaak zeldzame aandoeningen. Deze kinderarts heet kinderarts EAA: kinderarts voor erfelijke en aangeboren aandoeningen. In steeds meer ziekenhuizen in Nederland werken kinderartsen EAA. De kinderarts EAA stemt met de eigen kinderarts in de woonomgeving van het kind af hoe de zorg voor het kind zo optimaal mogelijk kan verlopen. Ook kan de kinderarts EAA gespecialiseerde kinderartsen om hulp vragen zoals een kinderMDL-arts of kinderchirurg voor darmproblemen, een kindernefroloog of een kinderneuroloog voor kinderen met epilepsie of bewegingsstoornissen.

Fysiotherapie
De kinderfysiotherapeut kan door middel oefeningen en advies aan ouders de ontwikkeling van het kind zo goed mogelijk stimuleren. Door veel te oefenen, leren kinderen hun hoofd op te tillen, te rollen, kruipen en te staan en lopen. Ook kan de kinderfysiotherapeut oefeningen geven om het evenwicht te trainen.

Logopedie
Een logopediste kan tips en adviezen geven indien er problemen zijn met zuigen, drinken, kauwen of slikken. Sommige kinderen hebben baat bij een speciale speen (special need speen) waardoor het drinken uit de fles beter verloopt. Moeders kunnen borstvoeding kolven, zodat kinderen op deze manier toch borstvoeding als voeding kunnen krijgen via de fles.
Ook kan de logopediste helpen om de spraakontwikkeling zo goed mogelijk te stimuleren. Praten kan ook ondersteund worden door middel van gebaren of pictogrammen. Op die manier kunnen kinderen zich leren uitdrukken ook als ze nog geen woorden kunnen gebruiken. Sommige kinderen hebben baat bij een spraakcomputer.

Ergotherapie
Een kinderergotherapeut kan adviezen geven hoe kinderen in het dagelijks leven zo goed mogelijk om kunnen gaan met hun evenwichtsproblemen. Ook kan de ergotherapeut advies geven over hulpmiddelen die er voor kunnen zorgen dat kinderen zo goed mogelijk kunnen functioneren.

Revalidatiearts
De revalidatiearts coördineert de verschillende behandelingen en vervolgt de ontwikkeling van kinderen met een ontwikkelingsachterstand. Bij problemen wordt gekeken wat voor oplossing er voor deze problemen te bedenken is. Vaak doet de revalidatiearts dit aan de hand van ICF-CY model. Er wordt gekeken wat het effect is van de aandoening op de verschillende lichaamsfuncties van het kind, de mogelijkheid om activiteit te ondernemen (bijvoorbeeld eten, aankleden, spelen) en de mogelijkheden om deel te nemen aan het dagelijks leven. De revalidatiearts denkt samen met een team mee welke oplossingen er te bedenken zijn voor een bepaald probleem.
De revalidatiearts geeft ook adviezen voor aangepaste schoenen of het gebruik van bijvoorbeeld spalken.
De revalidatiearts geeft ook adviezen voor het juiste onderwijs van het kind. Sommige kinderen gaan naar de school die verbonden is aan het revalidatiecentrum.
Ook voor jongere kinderen bestaan op het revalidatiecentrum vaak groepjes waarin de kinderen gedurende een dagdeel therapie krijgen waarin hun ontwikkeling gestimuleerd wordt.
Dagopvang
Vanaf de leeftijd van 2 maanden kunnen kinderen die niet naar een reguliere kinderdagopvang kunnen, naar een speciale kinderdagopvang toe gaan. Er bestaat speciale therapeutische peutergroepen in revalidatiecentra, of dagopvang in een orthopedagogisch dagcentrum (ODC) of in een medische kinderdagcentrum (MKD). Het hangt van de problemen die het kind ervaart af (zoals epilepsie of gedragsproblemen), welke vorm van dagopvang het meest geschikt is. Aanmelding voor een ODC of een MKD verloopt via de gemeente (vaak cia het centrum jeugd en gezin, via het jeugdteam of via het sociaal wijkteam). Aanmelding voor een therapeutische peutergroep in een revalidatiecentrum verloopt via de revalidatiearts.

Onderwijs
Een deel van de kinderen met het CAMTA1-syndroom kan normaal onderwijs volgen. Vaak is er wel wat extra begeleiding nodig in de vorm van remedial teaching of ambulante begeleiding. Een ander deel van de kinderen volgt speciaal onderwijs. Het is belangrijk om goed te kijken naar de sterke kanten van het kind en het kind extra te helpen bij kanten waarmee het meer moeite heeft (zoals onthouden, werktempo). Het LWOE kan leerkrachten adviezen geven hoe kinderen met epilepsie op school het beste begeleid kunnen worden.
Kinder- en jeugdpsychiater
Een kinder-en jeugdpsychiater kan advies geven hoe om te gaan met gedragsproblemen zoals ADHD en/of autisme. Soms is het nodig om gedragsregulerende medicatie zoals methylfenidaat voor ADHD of risperidon of aripiprazol voor autisme te geven. Per kind moeten de eventuele voordelen van het gebruik van deze medicijnen worden afgewogen tegen de nadelen ervan. Wanneer uw kind het lastig vindt om medicijnen in te nemen, vindt u hier tips voor innemen medicijnen.
Aanvalsbehandeling epilepsie
De meeste epilepsieaanvallen gaan vanzelf over binnen enkele minuten. Omstanders hoeven dan niets te doen om de aanval te doen stoppen. Het is belangrijk om zo rustig mogelijk te blijven en het kind zo veel mogelijk met rust te laten.
Wanneer een aanval na 5 minuten nog niet vanzelf gestopt is, dan zal vaak geadviseerd worden om medicijnen te geven om een aanval te doen stoppen. De behandelende arts zal altijd aangeven welk tijdstip voor een bepaald kind het beste is. Medicijnen die gebruikt kunnen worden voor het stoppen van een aanval zijn diazepam rectiole (Stesolid®), midazolam neusspray, midazolam rectiole, lorazepam of clonazepam druppels.
Het effect van deze medicijnen ontstaat na enkele minuten. Nadien zal het kind meestal in slaap vallen, soms ook niet.

Behandeling epilepsie
Met behulp van medicijnen wordt geprobeerd om de epilepsieaanvallen zo veel mogelijk te voorkomen en het liefst er voor te zorgen dat er helemaal geen epilepsieaanvallen meer voorkomen. Soms lukt dit vrij gemakkelijk met een medicijn, maar bij een deel van de kinderen is het niet zo eenvoudig en zijn combinaties van medicijnen nodig om de epilepsie aanvallen zo veel mogelijk of helemaal niet meer te laten voorkomen.
Verschillende soorten medicijnen kunnen gebruikt worden om de epilepsie onder controle te krijgen. Er bestaat geen duidelijk voorkeursmedicijn. Medicijnen die vaak gebruikt worden zijn natriumvalproaat (Depakine ®), levetiracetam (Keppra ®), clobazam (Frisium ®) en zonisamide (Zonegran®). Wanneer uw kind het lastig vindt om medicijnen in te nemen, vindt u hier tips voor innemen medicijnen.
Bij een deel van de kinderen zal het niet lukken om de epilepsieaanvallen met medicijnen onder controle te krijgen. Er bestaan ook andere behandelingen die een goed effect kunnen hebben op de epilepsie, zoals een ketogeen dieet, een nervus vagusstimulator, of een behandeling met methylprednisolon. Ook een combinatie van deze behandelingen met medicijnen die epilepsie onderdrukken is goed mogelijk.

Behandeling ataxie
Er bestaan geen medicijnen die ataxie kunnen verminderen. Het trainen van de balans helpt om minder last te hebben van evenwichtsproblemen. Het gebruik van zwaarder bestek of een verzwaarde pen kan helpen om minder last te hebben van het trillen van de handen.
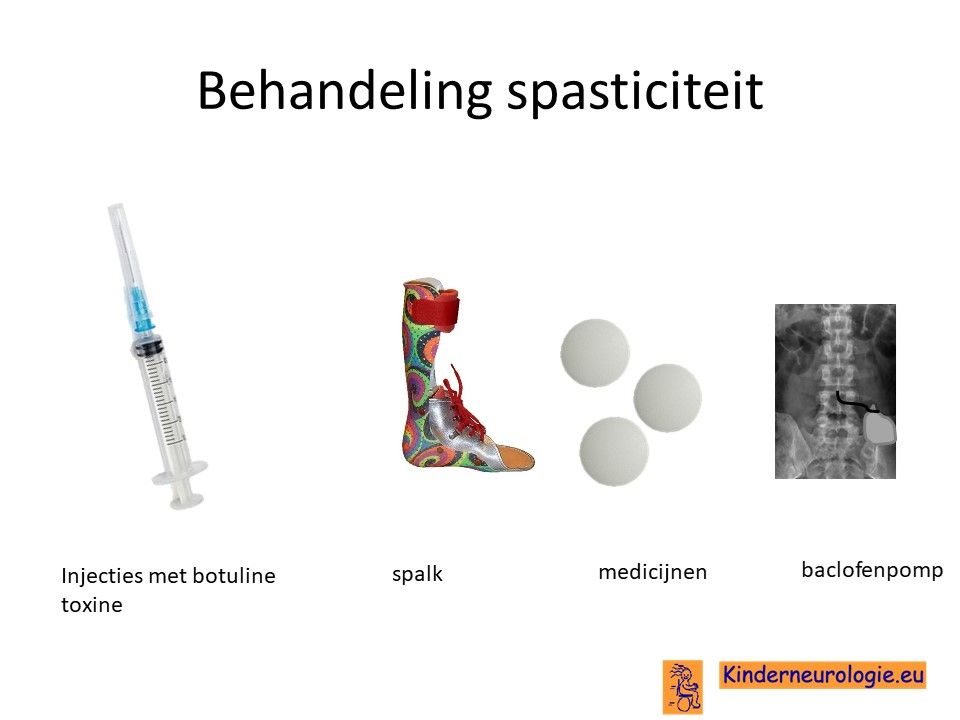
Behandeling spasticiteit
Injecties met botulinetoxine in een spastische spier kan de spasticiteit voor een aantal maanden verminderen. Met behulp van spalken en fysiotherapie kan op deze manier het looppatroon verbeterd worden. Vaak moeten deze injecties na een aantal maanden weer herhaald worden.
Ook kan met behulp van medicijnen geprobeerd worden om de spasticiteit van de benen te verminderen. Nadeel van al deze medicijnen is vaak dat ze de spierzwakte verergeren en in het hele lichaam effect hebben, niet alleen in de benen. Veel gebruikte medicijnen zijn baclofen (Lioresal ®) en trihexyfenidyl (Artane®). Baclofen kan ook in de vorm van een baclofenpomp worden toegediend.
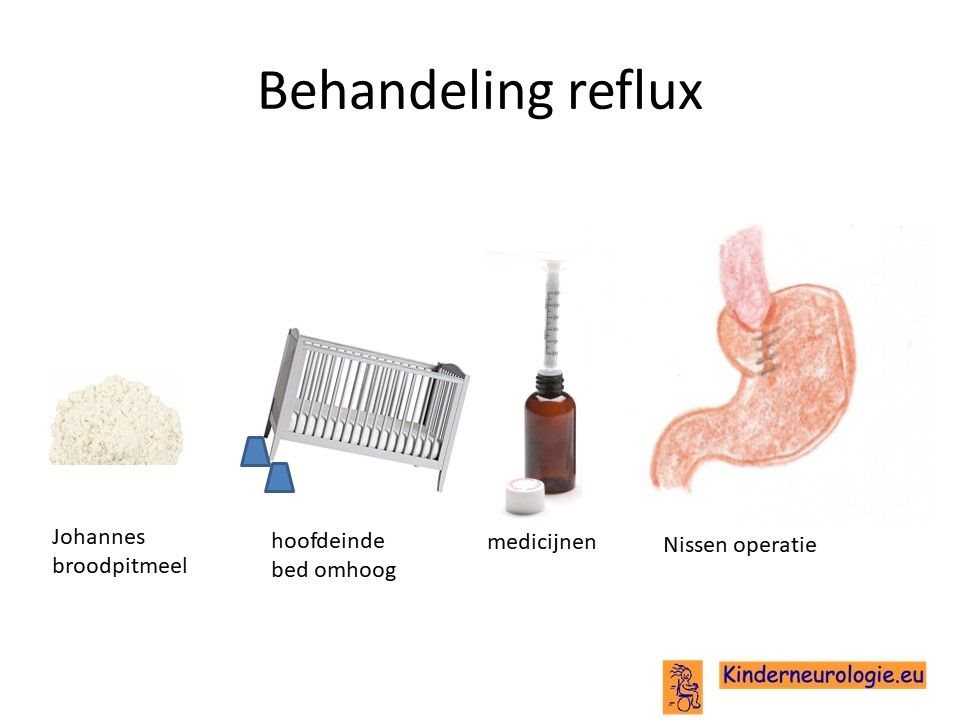
Reflux
Reflux kan er ook voor zorgen dat kinderen slecht eten. Door de voeding in te dikken met johannesbroodpitmeel kan de voeding minder gemakkelijk terug stromen van de maag naar de slokdarm. Ook zijn er medicijnen die de maaginhoud minder zuur kunnen maken waardoor de slokdarm minder geprikkeld wordt bij terugstromen van de maaginhoud. Medicijnen die hiervoor gebruikt worden zijn ranitidine, omeprazol of esomeprazol. Indien dit allemaal niet voldoende is, kan een operatie nodig zijn waarbij de overgang van de slokdarm naar de maag nauwer wordt gemaakt, waardoor de voeding ook minder gemakkelijk terug kan stromen. Dit wordt een Nissen-operatie genoemd.
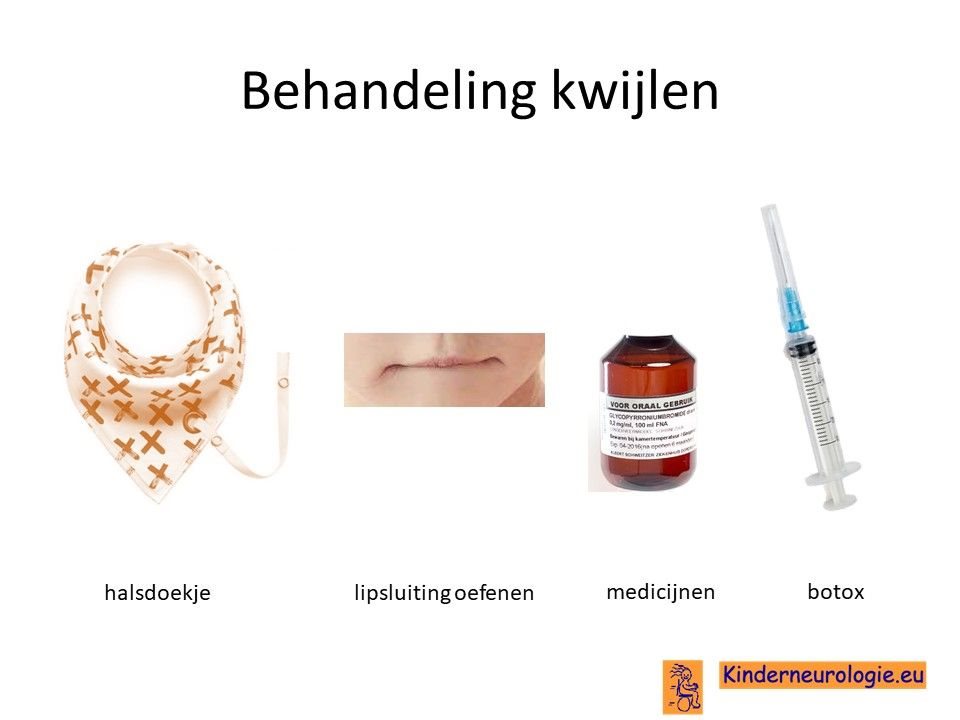
Kwijlen
Kwijlen kan verminderen door kinderen er bewust van te maken dat ze hun speeksel moeten doorslikken. Ook kunnen oefeningen waarbij geoefend wordt om de mond te sluiten helpen. Er bestaan moderne halsdoekjes die kwijl kunnen opvangen, zodat de kleding niet vies en nat wordt.
Er bestaan medicijnen die het kwijlen minder kunnen maken. Het meest gebruikte medicijn hierdoor is glycopyrrhonium. Soms kan een behandeling van de speekselklieren door middel van botox of door middel van een operatie nodig zijn om er voor zorgen dat kinderen minder kwijlen. Per kind zullen de voor- en nadelen van elke behandeling moeten worden afgewogen.
Verstopping van de darmen
Het medicijn macrogol kan er voor zorgen dat de ontlasting soepel en zacht blijft en stimuleert de darmwand om actief te blijven. Hierdoor kunnen kinderen gemakkelijker hun ontlasting kwijt. Verder blijft het belangrijk om te zorgen dat kinderen voldoende vocht en vezels binnen krijgen en zo veel als kan bewegen. Soms zijn zetpillen nodig om de ontlasting op gang te krijgen.

Zindelijkheid
Er kan met zindelijkheidstraining worden begonnen wanneer het kind zelf kan zitten op een potje en interesse begint te krijgen in het potje. Vaak is dit bij kinderen met dit syndroom op latere leeftijd dan gebruikelijk. Tips die kunnen helpen bij het zindelijk worden vindt u in de folder zindelijkheid.
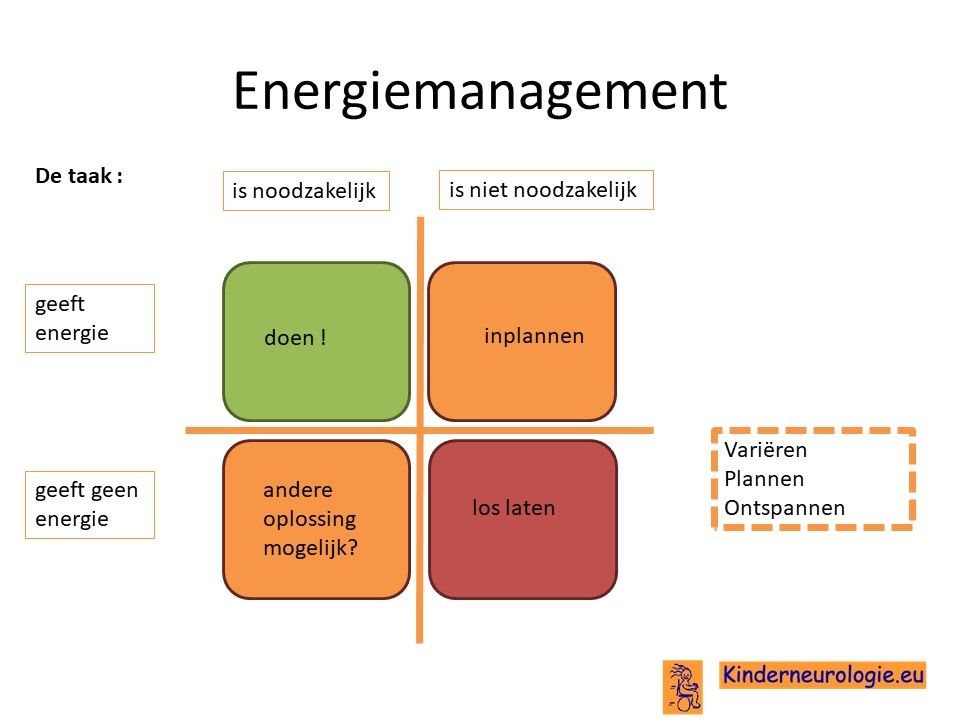
Omgaan met vermoeidheid
Het hebben van het CAMTA1-syndroom kost het lichaam bij alle dagelijkse activiteiten meer energie. Daardoor is de energie op een dag sneller op. Het goed verdelen van de energie over de dag helpt om vermoeidheid te voorkomen. Het kan helpen goed naar de taken die voor een dag ingepland staan te kijken en te beoordelen of deze taken noodzakelijk zijn of niet en of ze energie geven of niet. Aan de hand daarvan kan ingepland worden welke taken wel op het programma komen te staan en welke niet. Het maken van een planning kan helpen. Het is belangrijk om te zorgen voor variatie in taken en om ook voldoende momenten van ontspanning in te plannen.
Financiële kant van zorg voor een kind met een beperking
De zorg voor een kind met een beperking brengt vaak extra kosten met zich mee. Er bestaan verschillende wetten die zorg voor kinderen met een beperking vergoeden.
Daarnaast bestaan regelingen waar ouders een beroep op kunnen doen, om een tegemoetkoming te krijgen voor deze extra kosten. Meer informatie hierover vindt u in de folder financiën kind met een beperking.

Wat kun je als ouder zelf doen om de ontwikkeling van je kind optimaal te laten verlopen?
Bedenk dat wanneer je samen met je kind speelt, stoeit, danst, zingt, kletst, lacht en/of boekjes leest, dit ook allemaal manieren zijn waarop je kind zijn of haar hersenen traint om stappen voorwaarts te maken in de ontwikkeling. Het is dus niet zo dat alleen momenten van therapie, momenten van training zijn, wat veel ouders denken. Het is daarnaast goed om inspanning af te wisselen met ontspanning, dit is nodig om het geleerde te laten opslaan in de hersenen. De hele dag door training zonder rustmomenten, werkt juist averechts.
Daarnaast is het van onschatbare waarde je kind laten voelen dat je van hem of haar houdt, dat hij/zij geliefd is en zich mag ontwikkelen in een tempo die bij hem of haar past. Dit is extra van belang voor kinderen die zich anders ontwikkelen dan de "norm". "Goed zijn zoals je bent en gesteund te worden door mensen die van je houden is, heel belangrijk voor de ontwikkeling van een kind. Juist de ouders en de andere kinderen in het gezin die dichtbij het kind staan zijn daarin heel belangrijk om het kind daarin dit gevoel te geven. Het is goed dat ouders beseffen wat de waarde hiervan is voor het kind en welke rol zij hierin hebben.
Ook is het belangrijk om te bedenken wat goed voelt voor jullie als gezin en voor jou als ouder en waar jullie energie uithalen. Zorg ervoor dat er bewust ruimte is voor momenten die dit goede gevoel geven. Tot slot is het belangrijk dat je als ouders ook goed voor jezelf zorgt, de zorg voor een kind die zich anders ontwikkelt vraagt nog meer van ouders dan de zorg voor een kind die zich zonder problemen ontwikkelt. Het is goed om voor jezelf te zorgen of te laten zorgen, zodat je als ouder ook de energie houdt, om jouw kind te blijven begeleiden op een manier die bij jou past. Besef dat bij opvoeden hoort om te leren los laten. Veel ouders vinden dit lastig, zeker wanneer hun kind zich anders ontwikkelt dan andere kinderen. Maar dhet kan toch nodig zijn een deel van de zorg op bepaalde momenten uit handen te geven, ook als die ander het anders doet dan jij, je kind leert van deze verschillen en het geeft jou de mogelijk om zelf uit te rusten of nieuwe energie op te doen.

Wat kun je als gezin zelf doen om om te gaan met het hebben van een aandoening bij een gezinslid?
Als gezin van een kind waarbij er sprake is van een aandoening, is het goed om te zorgen dat jullie in de je kracht komen staan. Het is goed om te beseffen over welke denk-, emotionele-, innerlijke- en fysieke kracht jullie als gezin beschikken en hoe jullie deze kracht kunnen inzetten om goed voor ieder lid van het gezin te zorgen. Bekijk wat bij jullie als gezin past. Bekijk wat je kunt doen (of kunt laten) om deze kracht zo optimaal mogelijk in te zetten. En bedenk ook dat ieder lid van het gezin verschillende kwaliteiten heeft waarmee jullie elkaar kunnen aanvullen en kunnen versterken.
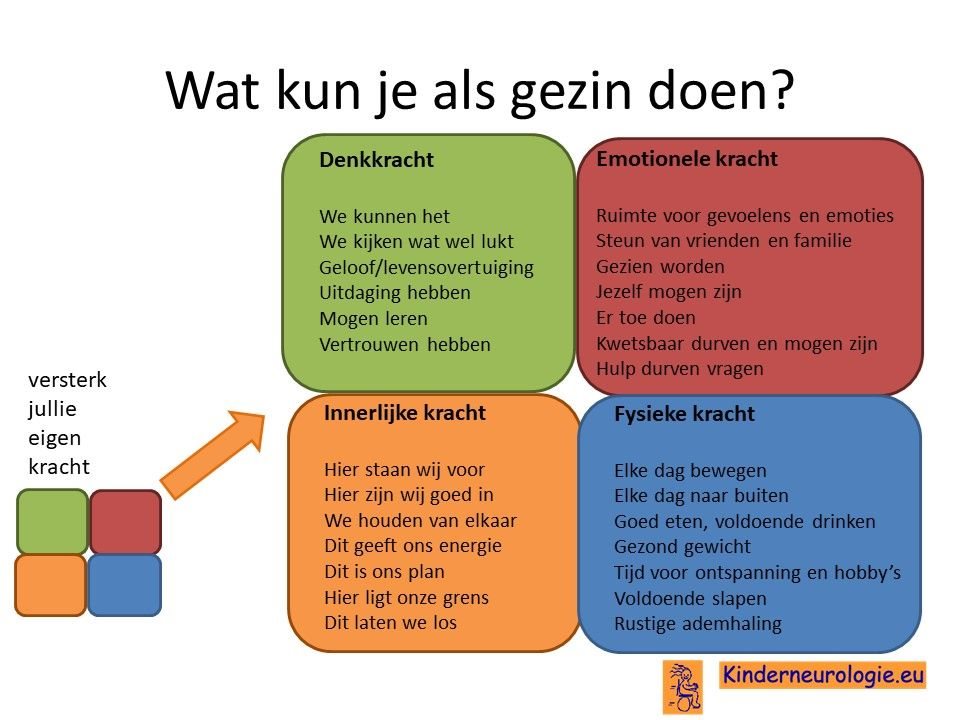
Begeleiding
Een maatschappelijk werkende of psycholoog kan ouders begeleiden om de aandoening van hun kind een plaats te geven in hun leven.Het kost vaak tijd om weer een nieuw evenwicht te vinden nadat ouders gehoord hebben dat er bij hun kind sprake is van een syndroom. Ook vinden veel ouders het vaak lastig hoe zij hun tijd en aandacht moeten verdelen tussen het kind met de beperking en andere kinderen in het gezin. In de folder aandacht en tijd voor brussen vindt u tips die u hierbij kunnen helpen.

Door het plaatsen van een oproepje op het forum van deze site kun u in contact komen met andere ouders die een kind hebben met het CAMTA1-syndroom of met een ander syndroom wat soortgelijke klachten geeft.
Wat betekent het hebben van het CAMTA1-syndroom voor de toekomst?
Geen duidelijke achteruitgang
De problemen die kinderen met het CAMTA-1 syndroom hebben lijken stabiel te blijven en niet toe te nemen in de loop van het leven.
Wel kan het zo zijn dat de problemen bij een jong kind nog niet zo opvallen, maar bij een ouder kind meer opvallen omdat van oudere kinderen meer verwacht wordt dan van jongere kinderen.
Zelfstandig leven
Een groot deel van de kinderen kan later als volwassenen wel zelfstandig leven. Een ander deel van de kinderen heeft daarbij blijvend hulp en ondersteuning nodig.
Ontwikkelingsmogelijkheden
Ouders willen vaak graag weten welke ontwikkelingsmogelijkheden hun kind heeft wanneer er sprake is van een verstandelijke beperking en of hun kind later in staat zal zijn een zelfstandig leven te leiden. Dit is heel moeilijk te voorspellen, zeker wanneer kinderen nog jong zijn, maar ook wanneer kinderen ouder zijn. Een van de factoren die een rol speelt bij de ontwikkelingsmogelijkheden is het IQ van het kind, maar daarnaast spelen ook andere factoren, zoals het wel of niet hebben van epilepsie, autisme, ADHD een rol. Het IQ zegt dus zeker niet alles, maar kan wel een bepaalde richting aangeven welke ontwikkelingsmogelijkheden reeel zijn om te verwachten. Dit is overigens wel een gemiddelde verwachting en dat betekent dat er altijd kinderen zijn die deze verwachting zullen behalen, maar dat dit voor anderen toch niet het geval blijkt te zijn of dat er juist meer ontwikkelingsmogelijkheden zijn dan er op grond van het IQ werd verwacht. Het blijft dus belangrijk naar het kind zelf te kijken en te bedenken dat pas op een bepaalde leeftijd duidelijk zal zijn wat het kind uiteindelijk allemaal in staat zal zijn om te leren.
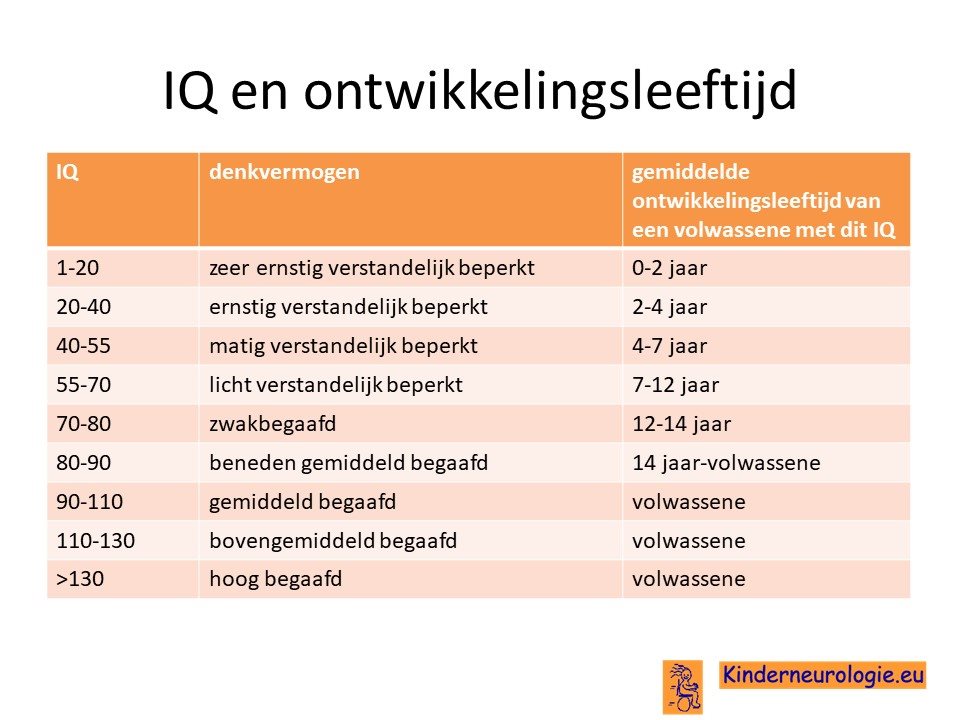
Transitie van zorg
Tussen de leeftijd van 16 en 18 jaar wordt de zorg vaak overgedragen van kinderspecialisten naar specialisten die de zorg aan volwassenen geven. Het is belangrijk om tijdig hierover na te denken. Is er behoefte de zorg over te dragen naar specialisten voor volwassenen of kan de huisarts de zorg leveren die nodig is.En als er behoefte is aan overdragen van de zorg naar specialisten voor volwassenen, naar welke dokter(s) wordt de zorg dan overgedragen? In welk ziekenhuis kan de zorg het beste geleverd worden. Het proces van overdragen van de zorg wordt transitie genoemd. Het is belangrijk hier tijdig over na te denken en een plan voor te maken samen met de dokters die betrokken zijn bij de zorg op de kinderleeftijd.
Ook is het belangrijk om te gaan werken aan zelfstandigheid van de jongere. Op de kinderleeftijd zorgen ouders dat de zorg rondom de jongere goed geregeld wordt, op volwassen leeftijd wordt dit steeds meer van de jongeren zelf verwacht. Het is goed om tijdig na te gaan op welke vlakken de jongere al zelfstandig is en op welke vlakken er getraind moet worden voor meer zelfstandigheid. De groeiwijzer kan hierin behulpzaam zijn.
Ook verandert er veel in de zorg wanneer een jongere de leeftijd van 18 jaar bereikt. Voor meer informatie over deze veranderingen verwijzing wij u naar het artikel veranderingen in de zorg 18+.
![]()
Arts VG
Een Arts VG is een arts die zich gespecialiseerd heeft in de zorg voor mensen met een verstandelijke beperking. De Arts VG richt zich op het voorkomen, behandelen en beperken van lichamelijke en psychische problemen die te maken hebben met een verstandelijke of lichamelijke beperking. De Arts VG werkt hiervoor samen met de huisarts, de medische specialist, de gedragsdeskundige en/of andere therapeuten (zoals een fysiotherapeut of een logopedist). Er zijn steeds meer poliklinieken in Nederland waar een Arts VG werken en waar kinderen en volwassenen met een verstandelijke beperking terecht kunnen met hun hulpvragen die te maken hebben met hun beperking. Daarnaast werken Artsen VG ook in instellingen en zijn ze betrokken bij gespecialiseerde kinderdagcentra. Op de website van de VGN is een lijst met poliklinieken te vinden waar Artsen VG werken.

Relaties
Voor volwassenen met een beperking kan het leggen en behouden van een vriendschap of een relatie met een ander meer moeite kosten dan voor volwassenen zonder beperking. Het gaat minder vanzelfsprekend omdat de volwassene bijvoorbeeld minder energie heeft, het lastiger vindt om zelf contacten te leggen, onzeker is, andere volwassenen niet goed weten hoe met een volwassene met een beperking om te gaan of omdat uitgaansgelegenheden minder goed toegankelijk zijn voor een volwassene met een beperking. Vaak ronden volwassenen hun opleiding af, zodat contact die voorheen via school met klasgenoten plaats vonden, niet meer vanzelfsprekend zijn. Voor een deel van de volwassenen verlopen nieuwe vriendschappen daarna via werk of de buurt waarin ze wonen. Sport is vaak een mooie manier om nieuwe vriendschappen op te doen. Via de website van uniek sporten, zijn adressen te vinden van sportmogelijkheden voor mensen met een beperking. Ook een hobby kan een mooie manier zijn om nieuwe contacten te leggen. Een ander deel van de volwassen vindt nieuwe vrienden via social media.
Werk
Een deel van de volwassenen met het CAMTA1-syndroom kan op volwassen leeftijd regulier werk uitvoeren. Het is niet verplicht de werkgever op de hoogte te stellen dat de diagnose CAMTA1-syndroom is gesteld. Het kan wel fijn zijn dat de werkgever op de hoogte is, zodat er bijvoorbeeld aanpassingen aan de werkplek worden gedaan of een mogelijkheid om een rustmoment gedurende de dag te nemen. De bedrijfsarts kan behulpzaam zijn bij het adviseren voor aanpassingen waardoor de volwassene met XXX op een goede en gezonde manier zijn werk kan doen.
Voor een ander deel van de volwassenen zal het niet mogelijk zijn om regulier werk te vinden. Zij kunnen een beroep doen op de participatiewet. Hiervoor kunnen volwassenen contact opnemen met de gemeente van de plaats waar zij wonen. De gemeente kijkt samen met de volwassene welke ondersteuning de volwassene nodig heeft om passend werk te vinden. Jobcoaches kunnen helpen bij het vinden van passend werk.
Rijbewijs
Het hebben van een lichamelijke en/of verstandelijke beperking kan van invloed zijn op de rijvaardigheid. Er zijn regels bij welke beperkingen een volwassene wel een auto mag besturen en bij welke beperkingen niet. Aanpassingen in de auto kan maken dat een volwassene wel een auto mag besturen. Op de eigen verklaring van het CBR moet ingevuld worden dat er sprake is van het CAMTA1-syndroom. Dit zal er vaak voor zorgen dat er een medische keuring nodig zal zijn. Soms wordt er ook een rijvaardigheidstest afgenomen. Het is mogelijk dat de geldigheidsduur van het rijbewijs korter is dan gebruikelijk vooral wanneer er sprake is van medische problemen die kunnen toenemen in de loop van de tijd. Meer informatie over het rijbewijs is te vinden op de website van het CBR.

Vermoeidheid
Volwassenen met het CAMTA1-syndroom zijn vaak sneller vermoeid dan volwassenen zonder het CAMTA1-syndroom. Dit vraagt vaak aanpassing in het dagelijks leven. Zorgen voor een vast dagritme waarin activiteiteiten worden afgewisseld met momenten van rust en ontspanning helpt om de energie goed over de dag te verdelen. Ook is het belangrijk elke dag lichamelijk actief te zijn en te zorgen voor een goede conditie. Daarnaast zijn vaste tijden van gaan slapen in een koele donker kamer en vaste tijden van wakker worden belangrijk om te zorgen voor voldoende goede slaap.
Vaak moet er een keuze gemaakt worden welke activiteiten op een dag ingepland gaan worden. Het is goed om te kijken of deze activiteiten noodzakelijk zijn om zelf te doen of niet(wellicht kan iemand anders deze taak overnemen?) en of ze wel of geen energie geven. Op deze manier kan bepaald worden welke activiteiten op een dag het beste ingepland kunnen worden.
Levensverwachting
Er zijn geen aanwijzingen dat de levensverwachting van kinderen met het CAMTA1-syndroom anders is dan die van kinderen zonder dit syndroom.
Kinderen krijgen
Het CAMTA1-syndroom is een erfelijke aandoening. Er zijn geen aanwijzingen dat het hebben van deze aandoening van invloed is op de vruchtbaarheid.. Kinderen van een volwassene met deze aandoening hebben 50% kans om zelf ook deze aandoening te krijgen. Het valt niet te voorspellen in welke mate kinderen last van deze aandoening zullen krijgen. Dit kan in dezelfde mate, in mindere mate of in ernstige mate zijn. Indien de volwassene geen kinderen wil of kan krijgen, moet wellicht nagedacht moeten worden over anticonceptie, waarover u in deze folder meer informatie vindt.
Hebben broertjes en zusjes een vergrote kans om het CAMTA1-syndroom te krijgen?
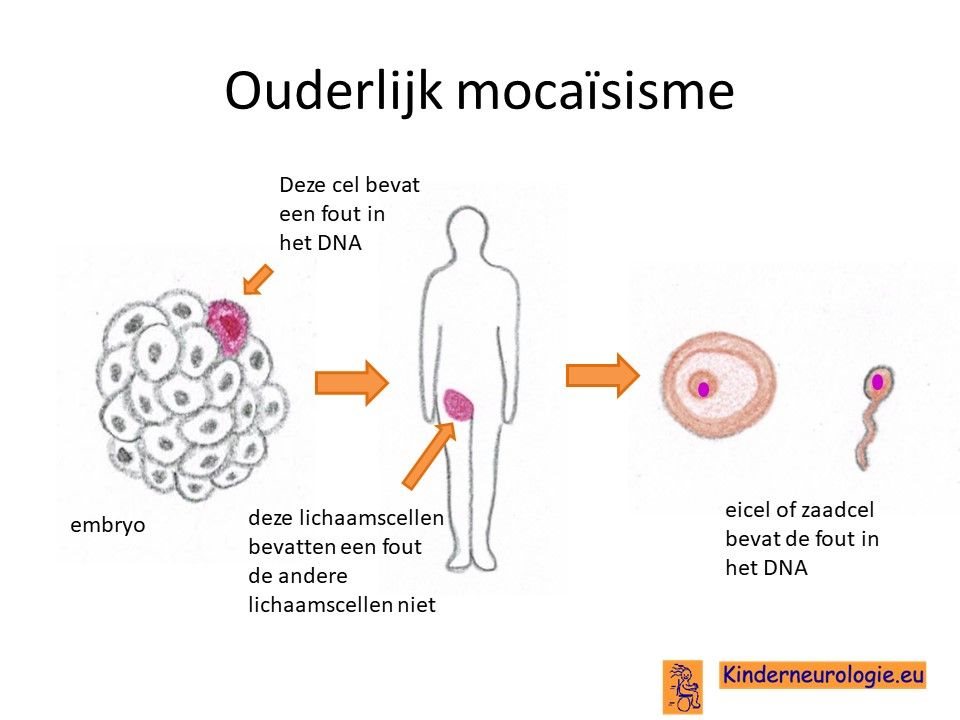
Het CAMTA1-syndroom is een erfelijke aandoening. Soms hebben kinderen het foutje geerfd van een van de ouders, in die situatie hebben broertjes en zusjes tot 50% kans om ook het CAMTA1-syndroom te krijgen. Het kan ook zijn dat het foutje bij het kind zelf ontstaan is, dan hebben broertjes en zusjes geen vergrote kans om ook het CAMTA1-syndroom te krijgen. Heel soms komt het voor dat de moeder in haar eicellen of de vader in zijn zaadcellen het foutje in het CAMTA1-gen heeft zitten, zonder dat dit in de rest van hun lichaam aanwezig is. In die situatie hebben zij zelf geen klachten, maar hebben broertjes en zusjes wel een verhoogde kans om zelf het CAMTA1-syndroom te krijgen.
Een klinische geneticus kan hier meer informatie over geven.
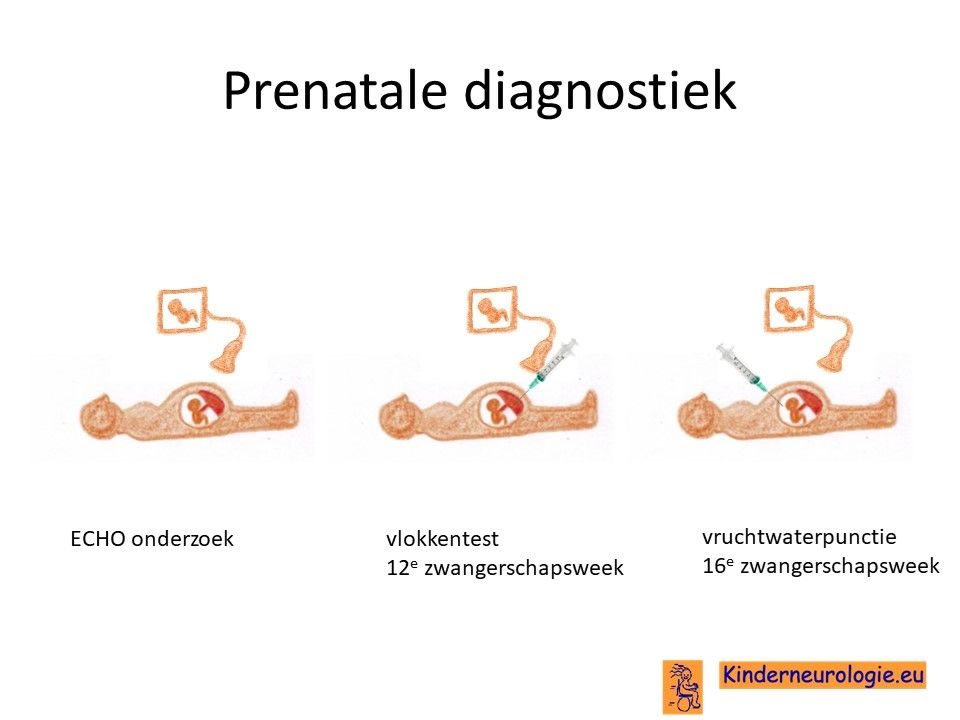
Prenatale diagnostiek
Door middel van een vlokkentest in de 12e zwangerschapsweek of een vruchtwaterpunctie in de 16e zwangerschapsweek bestaat de mogelijkheid om tijdens een zwangerschap na te gaan of een broertje of zusje ook deze aandoening heeft. Beide ingrepen hebben een klein risico op het ontstaan van een miskraam (0,5% bij de vlokkentest en 0,3% bij de vruchtwaterpunctie). De uitslag van deze onderzoeken duurt twee weken. Voor prenatale diagnostiek kan een zwangere de 8ste week verwezen worden door de huisarts of verloskundige naar een afdeling klinische genetica. Meer informatie over prenatale diagnostiek kunt u vinden op de website: www.pns.nl
Wilt u dit document printen dan kunt u hiervoor de printbutton bovenaan de pagina gebruiken
Wilt u ook uw verhaal kwijt, dat kan: verhalen kunnen gemaild worden via via contact en zullen daarna zo spoedig mogelijk op de site worden geplaatst. Voor meer informatie zie hier.
Heeft u foto's die bepaalde kenmerken van deze aandoening duidelijk maken en die hier op de website mogen worden geplaatst, dan vernemen wij dit graag via contact.
Links
www.ataxie.nl
(Site van patientenverenging voor mensen met ataxie)
Referenties
- Intragenic CAMTA1 rearrangements cause non-progressive congenital ataxia with or without intellectual disability. Thevenon J, Lopez E, Keren B, Heron D, Mignot C, Altuzarra C et al. J Med Genet. 2012;49:400-8.
- Neuropsychological and neuroimaging phenotype induced by a CAMTA1 mutation. Magnin E, Blagosklonov O, Sylvestre G, Minot D, Thevenon J, Faivre L, Boulahdour H, Thauvin-Robinet C, Rumbach L. Brain Dev. 2014;36:711-5.
- Intragenic CAMTA1 deletions are associated with a spectrum of neurobehavioral phenotypes. Shinawi M, Coorg R, Shimony JS, Grange DK, Al-Kateb H. Clin Genet. 2015;87:478-82.
- Novel Nonsense Calmodulin-Binding Transcription Activator 1 Mutation Presenting as a Tremor-Predominant Phenotype. Agarwal S, Gilbert R, Lau HA. Mov Disord Clin Pract. 2016;3:611-614
- Genetic diagnoses in epilepsy: The impact of dynamic exome analysis in a pediatric cohort. Rochtus A, Olson HE, Smith L, Keith LG, El Achkar C, Taylor A, Mahida S, Park M, Kelly M, Shain C, Rockowitz S, Rosen Sheidley B, Poduri A. Epilepsia. 2020;61:249-258.
- De novo variants in CAMTA1 cause a syndrome variably associated with spasticity, ataxia, and intellectual disability. Wijnen IGM, Veenstra-Knol HE, Vansenne F, Gerkes EH, de Koning T, Vos YJ, Tijssen MAJ, Sival D, Darin N, Vanhoutte EK, Oosterloo M, Pennings M, van de Warrenburg BP, Kamsteeg EJ. Eur J Hum Genet. 2020;28:763-769
- Expanding the molecular spectrum and the neurological phenotype related to CAMTA1 variants. Jacobs EZ, Brown K, Byler MC, D'haenens E, Dheedene A, Henderson LB, Humberson JB, van Jaarsveld RH, Kanani F, Lebel RR, Millan F, Oegema R, Oostra A, Parker MJ, Rhodes L, Saenz M, Seaver LH, Si Y, Vanlander A, Vergult S, Callewaert B. Clin Genet. 2021;99:259-268
- CAMTA1-related disorder: Phenotypic and molecular characterization of 26 new individuals and literature review. Al-Kateb H, Au PYB, Berland S, Cogne B, Demurger F, Fluss J, Isidor B, Frank LM, Varvagiannis K, Koolen DA, McDonald M, Montgomery S, Moortgat S, Deprez M, Karadurmus D, Paulsen J, Reis A, Rieger M, Vasileiou G, Willing M, Shinawi M. Clin Genet. 2024;105:294-301.
Laatst bijgewerkt: 2 oktober 2024 voorheen: 12 juli 2023, 8 juni 2022, 23 maart 2021, 7 maart 2020, 12 januari 2019 en 26 januari 2013
Auteur: JH Schieving