Wat is KCNMA1-syndroom?
KCNMA1-syndroom is een aandoening waarbij kinderen last hebben van aanvallen van epilepsie en/of van aanvallen met ongewilde bewegingen als gevolg van een fout in het erfelijk materiaal op de plaats van het KCNMA1-gen.
Hoe wordt KCNMA1-syndroom ook wel genoemd?
KCMA1-gen is de plaats in het erfelijk materiaal waar kinderen met deze aandoening een fout hebben. Een syndroom is een combinatie van symptomen die gezamenlijk voorkomen.
Paroxysmale non-kinesiogene dyskinesie type 3
Inmiddels is bekend dat een deel van de kinderen met het KCNMA1-syndroom aanvallen met ongewilde bewegingen heeft die paroxysmale non-kinesiogene dyskinesie worden genoemd. Er bestaan ook twee andere fouten in het DNA die deze aanvallen met ongewilde bewegingen kunnen geven, daarom wordt het type veroorzaakt door een fout in het KCNMA1-gen type 3 genoemd. Het woord paroxysmaal betekent aanvalsgewijs. Het woord non-kinesiogeen betekent dat de aanvallen niet uitgelokt worden door het maken van een beweging. Dysknisie is het medische woord voor onbedoelde bewegingen.
Liang-Wang syndroom
Liang en Wang zijn twee artsen die artsen die een syndroom met ontwikkelingsachterstand, balansproblemen en epilepsie als gevolg van een fout in het KCNMA1-gen hebben beschreven. Het wordt ook afgekort tot de letters LIWAS.
Spectrum
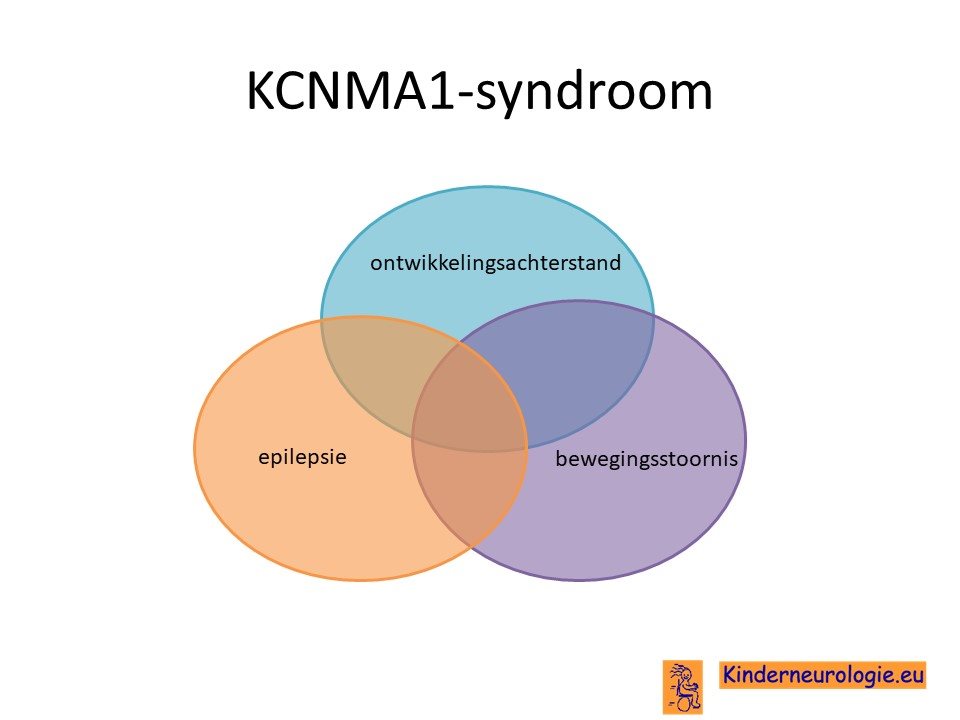
Kinderen met een fout in het KCNMA1-gen kunnen verschillende beelden laten zien. Een deel van de kinderen heeft met name last van een problemen met bewegen. Een ander deel van de kinderen heeft last van epilepsie als gevolg van het foutje in het KCNMA1-syndroom. Er zijn ook kinderen die zowel last van de epilepsie als van de bewegingsstoornis hebben. Een deel van de kinderen heeft een ontwikkelingsachterstand, een ander deel van de kinderen niet. Het KCNMA1-syndroom is dus een spectrum waarbij kinderen een of meerdere problemen kunnen hebben.
Hoe vaak komt KCNMA1-syndroom voor bij kinderen?
KCNMA1-syndroom is een zeldzaam voorkomende aandoening. Het is niet goed bekend hoe vaak deze aandoening bij kinderen voorkomt. Geschat wordt dat deze aandoening bij minder dan één op de 100.000 kinderen voorkomt. Wereldwijd zijn momenteel 50 mensen met het KCNMA1-syndroom bekend. Het is goed mogelijk dat er meer kinderen en volwassenen zijn die nog niet de juiste diagnose hebben gekregen.

Bij wie komt KCNMA1-syndroom voor?
KCNMA1-syndroom is al voor de geboorte aanwezig. De eerste klachten kunnen op verschillende leeftijd ontstaan. De kinderen die tot nu toe bekend zijn met het KCNMA1-syndroom hebben klachten gekregen tussen de leeftijd van een aantal maanden en de leeftijd van 16 jaar. Het is ook mogelijk dat er in de toekomst volwassenen ontdekt worden, die pas op volwassen leeftijd voor het eerst klachten krijgen.
Zowel jongens als meisjes kunnen het KCNMA1-syndroom krijgen.
Wat is de oorzaak van een KCNMA1-syndroom?
Fout in erfelijk materiaal
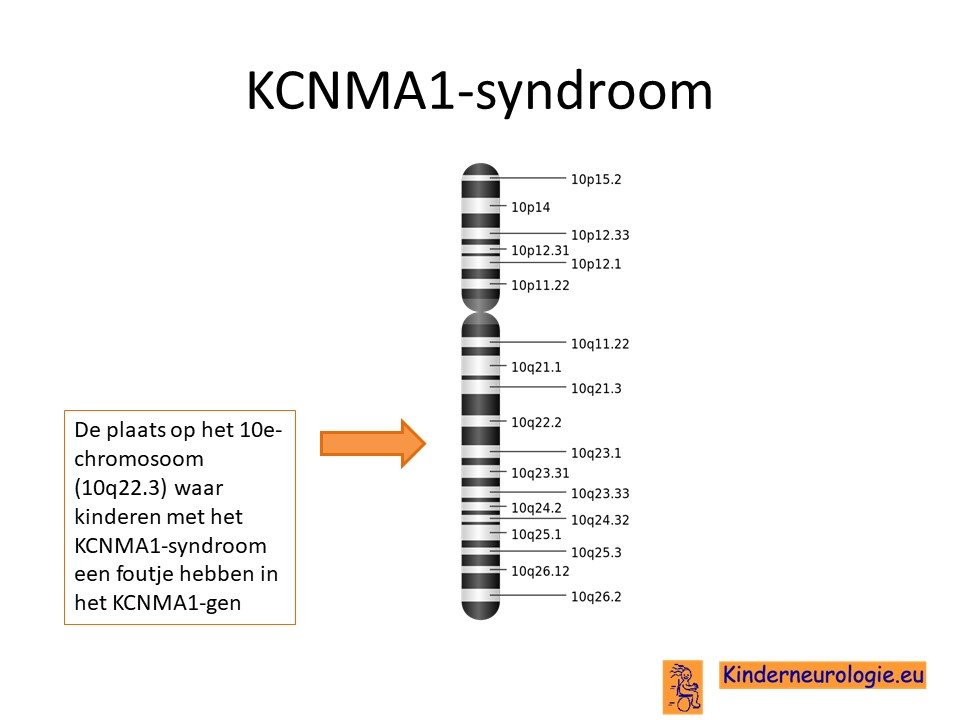
KCNMA1-syndroom wordt veroorzaakt door een foutje in het erfelijk materiaal van chromosoom 10 op een plaats die het KCNMA1-gen wordt genoemd.
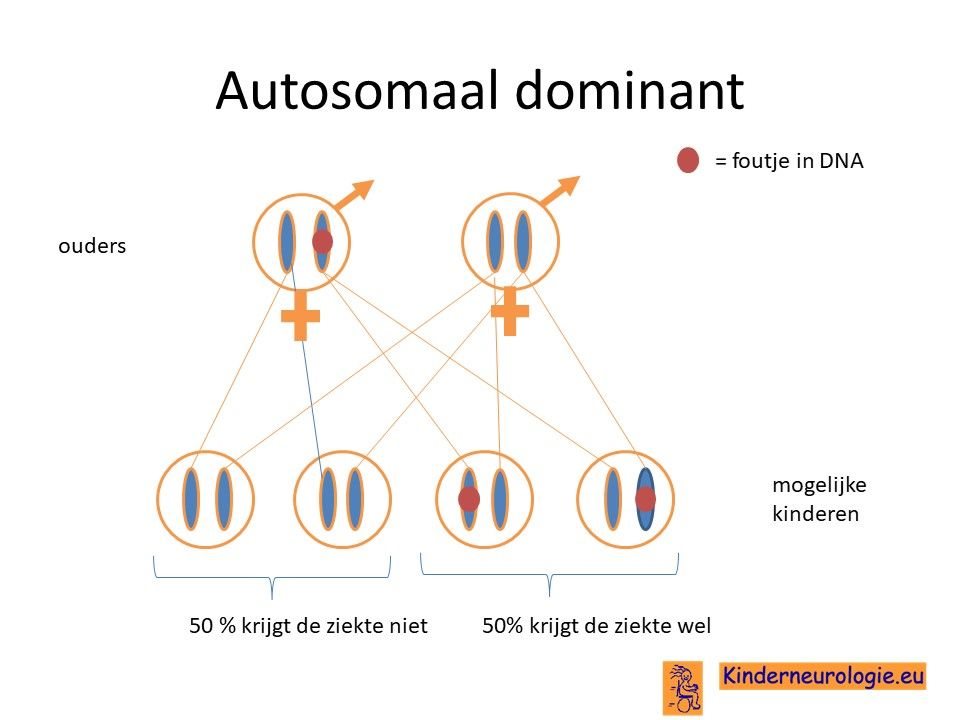
Autosomaal dominant
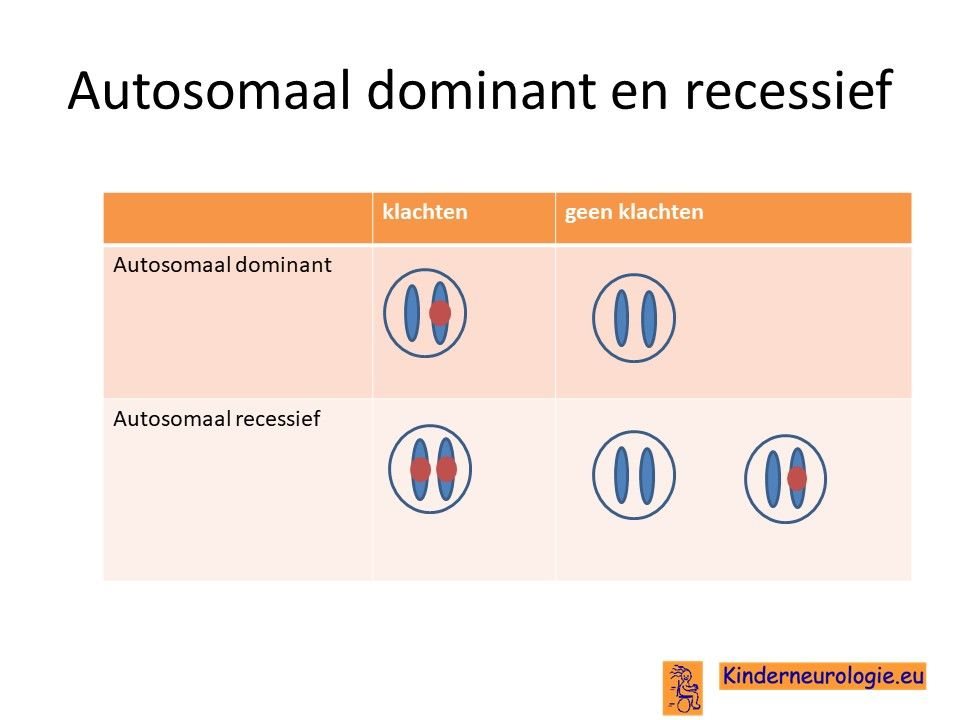
KCNMA1-syndroom is een zogenaamde autosomaal dominant fout, dat wil zeggen dat een fout op een van de twee chromosomen 10 die een kind heeft in het KCNMA1-gen al voldoende is om de aandoening te krijgen. Dit in tegenstelling tot een autosomaal recessieve fout waarbij kinderen pas klachten krijgen wanneer beide chromosomen een fout bevatten.
Bij het kind zelf ontstaan
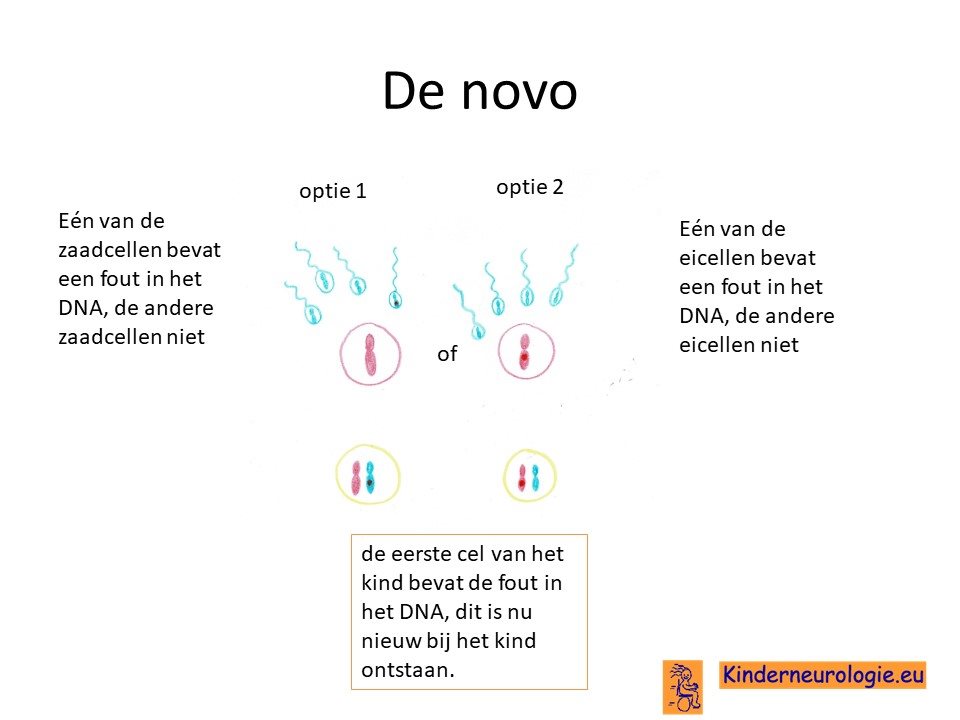
Bij een deel van de kinderen is het foutje in het erfelijk materiaal bij het kind zelf ontstaan bij de bevruchting van de eicel met de zaadcel. Nieuw ontstaan bij een kind wordt ook wel de novo genoemd.
Geërfd van een ouder
Een deel van de kinderen met het KCNMA1-syndroom heeft de fout in het DNA geërfd van een ouder. Soms was al bekend dat de ouder ook last heeft van epilepsie, of een bewegingsstoornis maar het kan ook zijn dat de ouder nooit klachten heeft gehad.
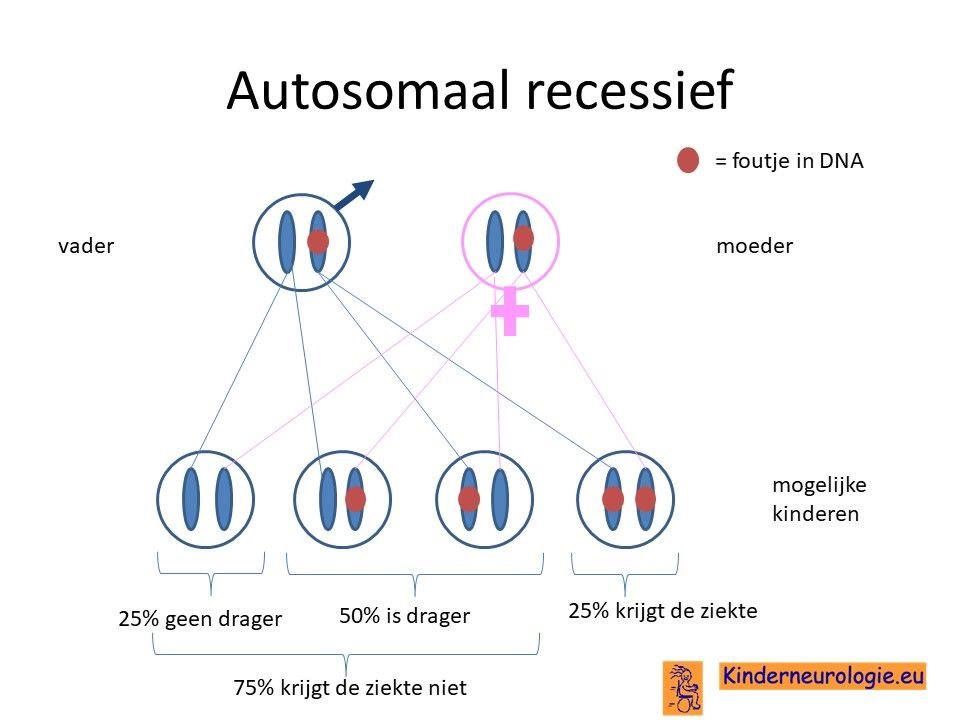
Autosomaal recessieve vorm
Er zijn inmiddels ook kinderen bekend die op beide chromosomen 10 een foutje hebben in het KCNMA1-gen. Zij hebben vaak een ernstiger ziektebeeld met epilepsie, een ontwikkelingsachterstand en balansproblemen. Bij deze kinderen wordt gesproken van een autosomaal recessieve vorm.
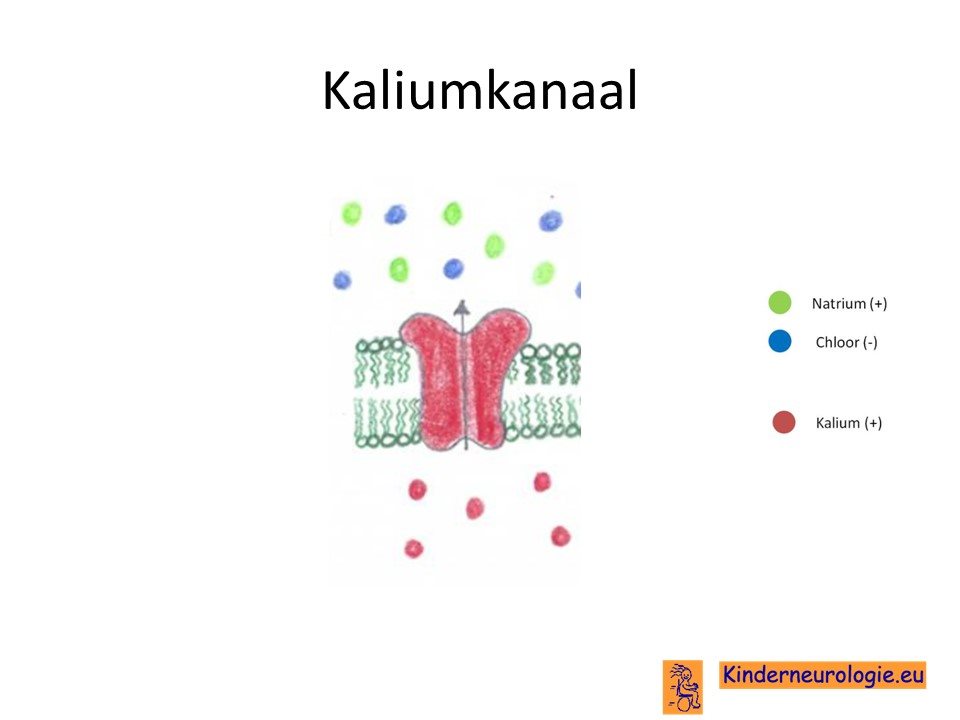
Kaliumkanaal
Het KCNMA1-gen bevat informatie voor de bouw van een kaliumkanaal in de hersencellen. Het gaat om een speciaal kaliumkanaal wat actief wordt wanneer er calcium in de buurt aanwezig is en wordt een zogenaamd BK-kanaal genoemd. Dit kaliumkanaal speelt een belangrijke rol bij de elektrische geleiding in de hersencellen. Wanneer dit kaliumkanaal overactief werkt, dan verloopt de elektrische geleiding in de hersencellen anders dan zou horen. Hersencellen kunnen dan spontaan overactief worden en signalen aan elkaar door geven. Dit is de oorzaak voor het ontstaan van de epilepsie of de bewegingsstoornis.
Spieren
Dit kaliumkanaal speelt ook een belangrijke rol in de spieren van het lichaam. Het zorgt er voor dat de spieren kunnen aanspannen.
Gain of loss of function
Bepaalde fouten in het DNA zorgen er voor dat het kaliumkanaal harder gaat werken dan gebruikelijk, terwijl andere fouten in het DNA er voor zorgen dat het kaliumkanaal minder hard gaat werken dan gebruikelijk. Het harder werken wordt gain of function genoemd, het minder hard werken loss of function.
Wanneer er sprake is van een gain of function ontstaat vaak epilepsie of aanvallen van een bewegingsstoornis. Bij het Liang-Wang syndroom is er juist vaak sprake van loss of function.

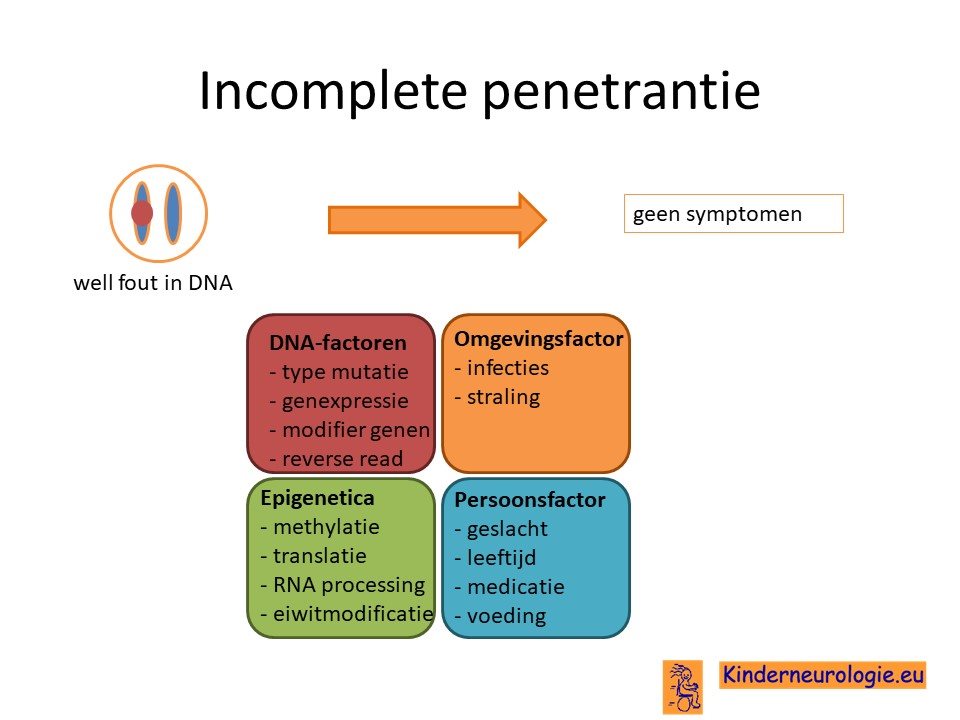
Incomplete penetrantie
Niet ieder persoon die een foutje heeft in het KCNMA1-gen krijgt ook daadwerkelijk klachten als gevolg van het hebben van dit foutje. Sommige mensen krijgen nooit tijdens hun leven klachten. Dit wordt ook wel incomplete penetrantie genoemd.
Wat zijn de symptomen van een KCNMA1-syndroom?
Variatie
Er bestaat een grote variatie in hoeveelheid en in ernst van de symptomen die verschillende kinderen met een KCNMA1-syndroom hebben. Dit valt van te voren niet goed te voorspellen.
Ook is er nog maar weinig bekend over het KCNMA1-syndroom. Het is goed mogelijk dat er andere symptomen horen bij deze aandoening die hier nog niet beschreven zijn.

Zwangerschap en geboorte
Tijdens de zwangerschap zijn er meestal geen bijzonderheden. Kinderen met een KCNMA1-syndroom worden na een normale zwangerschapsduur geboren en hebben een normaal geboortegewicht en lengte. Na de geboorte zijn er meestal geen bijzonderheden. De eerste levensmaanden verlopen normaal.
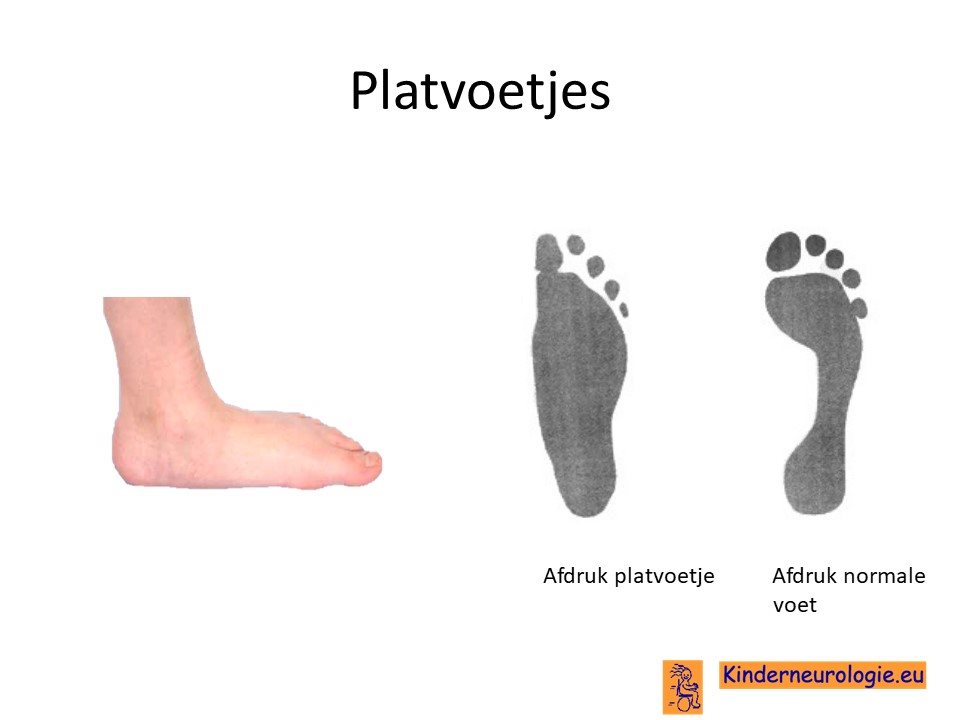
Lage spierspanning
Een deel van de kinderen met het KCNMA1-syndroom heeft een lage spierspanning. Hierdoor kunnen gewrichten gemakkelijker overstrekt worden. De lage spierspanning zorgt vaak voor platvoeten.
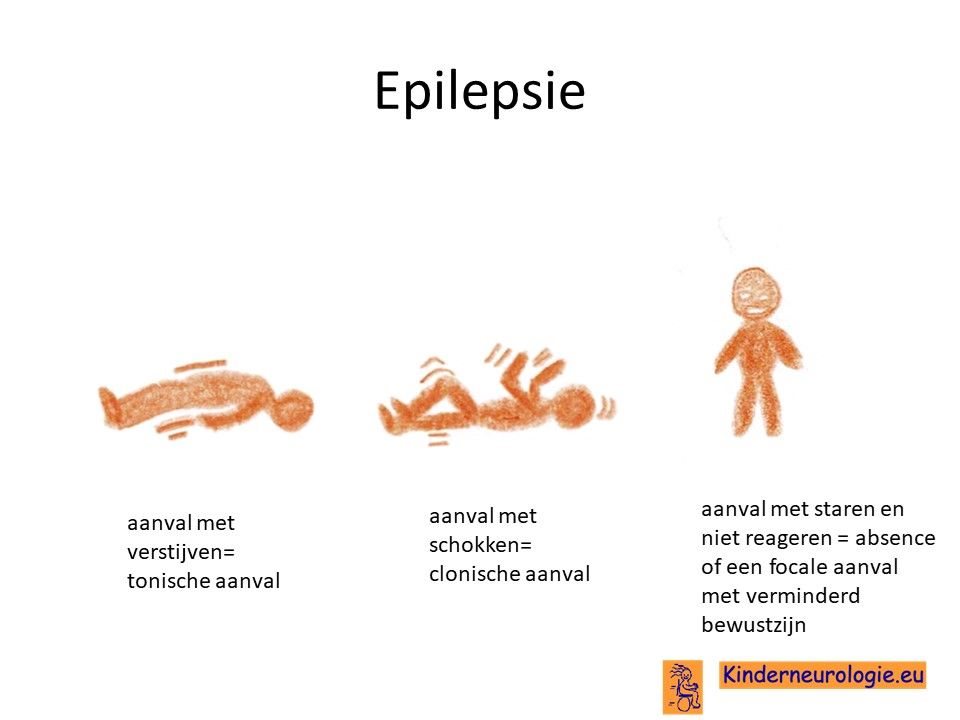
Epilepsie
Een deel van de kinderen met het KCNMA1-syndroom krijgt last van epilepsieaanvallen. De epilepsie kan op verschillende leeftijden ontstaan. Kinderen die op jonge leeftijd last krijgen van epilepsieaanvallen hebben vaak meer last van epilepsieaanvallen, dan kinderen die pas op oudere leeftijd last krijgen van epilepsieaanvallen. Verschillende soorten epilepsieaanvallen kunnen ontstaan zoals aanvallen met schokjes (myoclonieën genoemd), aanvallen met verstijven (tonische aanvallen), aanvallen met verslappen (atone aanvallen), aanvallen met verstijven en schokken (tonisch-clonische aanvallen) of aanvallen met staren en geen contact krijgen (absences genoemd). Kinderen kunnen meerdere type aanvallen hebben. Ook kan het zo zijn dat kinderen met het ouder worden, ander type epilepsieaanvallen ontwikkelen.
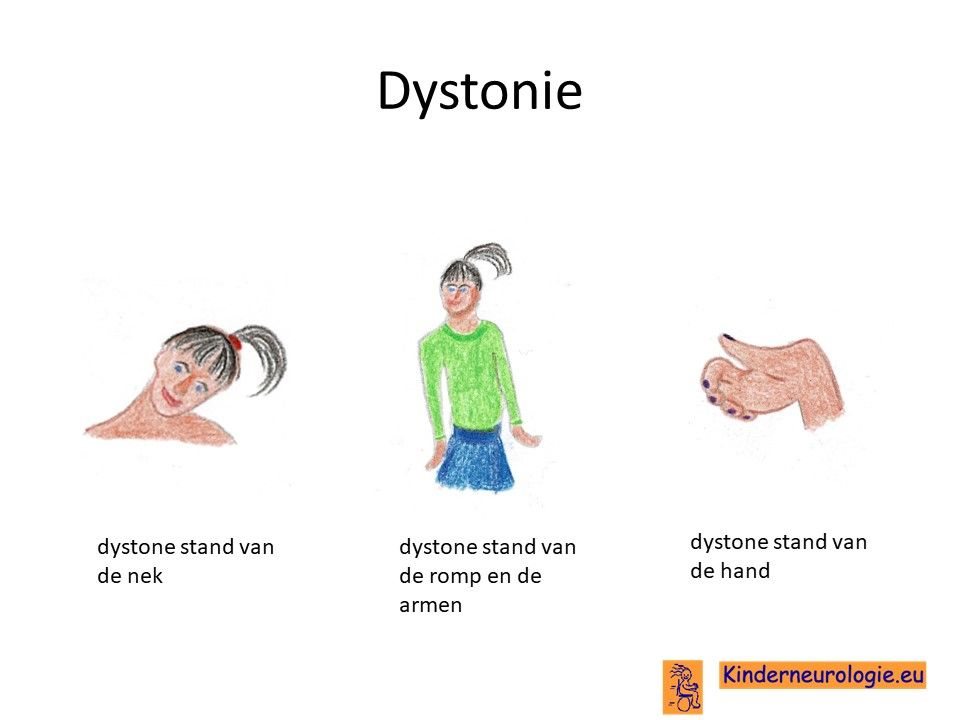
Bewegingsstoornis
Een ander deel van de kinderen met het KCNMA1-syndroom krijgt aanvallen van onbedoelde bewegingen. Dit kan veel lijken op epilepsieaanvallen, maar het verschil met epilepsie is, dat kinderen bij deze aanvallen bij bewustzijn zijn en de aanvallen bewust mee maken. Tijdens de epilepsieaanvallen zijn kinderen buiten bewustzijn. Tijdens de aanvallen van de bewegingsstoornis maken de armen en benen onrustige schokkerige, draaiend en strekkende bewegingen. Dit wordt ook wel dyskinesie genoemd. Soms wordt er een meer specifieke naam aangegeven zoals chorea of dystonie. Kenmerkend voor deze aandoening zijn ook afwijkende bewegingen met de mond, dit worden orofaciale dyskinesieen genoemd. De mond valt vaak open en kan niet dicht gedaan worden door het kind. De aanvallen houden vaak seconden tot minuten aan en verdwijnen dan weer spontaan. Tussen de aanvallen in bewegen kinderen normaal. De aanvallen kunnen worden uitgelokt door vermoeid of veel stress. Sommige kinderen hebben tientallen van deze aanvallen per dag, wat grote invloed heeft op de ontwikkeling van een kind.
Drop attacks
Een kenmerkend type aanval voor een deel van de kinderen met het KCNMA1-syndroom zijn aanvallen die ook wel drop attakcs genoemd. Tijdens deze aanvallen vallen kinderen plotseling op de grond zonder aanleiding. Kinderen zijn tijdens dit vallen gewoon bij bewustzijn, maar zijn niet in staat om te reageren. Tijdens dit neervallen kunnen de spieren van de armen en benen aangespannen of helemaal slap zijn waardoor kinderen hun armen en benen niet kunnen gebruiken om zich zelf op te vangen. Dit tijdelijk niet kunnen bewegen wordt akinesie genoemd. Soms maken de mond of de tong een vreemde beweging tijdens deze aanvallen. De aanvallen duren vaak 10-20 seconden en kunnen meerdere keren per dag voorkomen. Deze aanvallen worden nog al eens verward met epilepsieaanvallen.
Problemen met het evenwicht
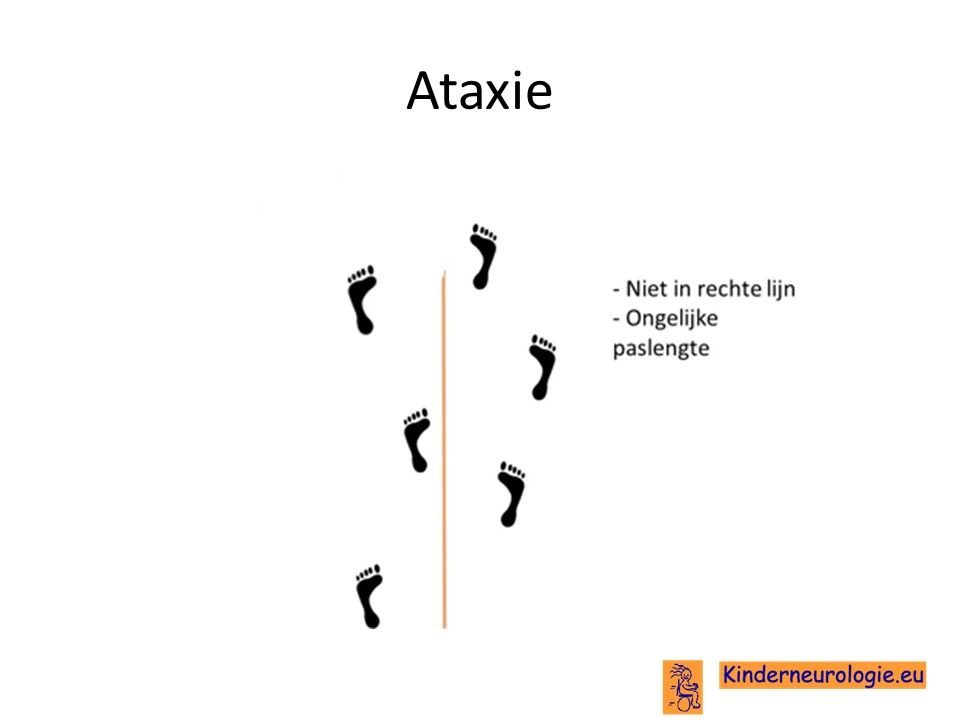
Het is voor kinderen met verlies van de functie van het BK-kanaal vaak lastiger om hun evenwicht te bewaren. Ze vallen gemakkelijker dan andere kinderen. Vaak zetten kinderen hun voeten wat verder uit elkaar om zo meer steun te hebben en minder snel om te vallen. Dit wordt een breedbasisch looppatroon genoemd. Kinderen zetten de ene keer een te grote pas en de andere keer juist een kleine pas. De problemen met het evenwicht wordt ook wel ataxie genoemd.
De handen kunnen een trillende beweging maken wanneer kinderen wat willen pakken. Dit wordt een tremor genoemd.
Daardoor wordt het bijvoorbeeld moeilijker om te schrijven, een kopje naar de mond te brengen of knoopjes dicht te maken.
Problemen met zien
Kinderen met verlies van functie van het BK-kanaal kunnen problemen zien hebben. Zij zien wazig.
Problemen met horen
Kinderen met verlies van functie van het BK-kanaal kunnen problemen horen hebben. Zij kunnen slechthorend zijn.

Ontwikkeling
Een deel van de kinderen met het KCNMA1-syndroom ontwikkelt zich normaal tussen de aanvallen door. Dit geldt vooral voor kinderen die weinig last hebben van aanvallen. Deze groep kinderen leert net als andere kinderen te zitten, te staan, te lopen en te praten.
Kinderen die veel last hebben van epilepsieaanvallen ontwikkelen zich vaak trager dan andere kinderen. Het hebben van deze aanvallen kost veel energie en deze energie kan niet in de ontwikkeling gestoken worden. Het kost deze kinderen meer tijd om te leren staan, lopen en praten. Zij leren dit vaak op een latere leeftijd dan leeftijdsgenoten die geen KCNMA1-syndroom hebben. Bij hen is er sprake van een ontwikkelingsachterstand.
Autistiforme kenmerken
Kinderen met een KCNMA1-syndroom hebben vaker autistiforme kenmerken. Ze zijn meer in zich zelf gekeerd en hebben niet zo’n behoefte aan contact met andere mensen. Het maken van oogcontact vinden kinderen vaak moeilijk.
Kinderen met autistiforme kenmerken houden vaak van een vaste voorspelbare structuur in de dag. Zij vinden het lastig wanneer hiervan wordt afgeweken. Ook onverwachte gebeurtenissen zijn moeilijk. Kinderen kunnen door onverwachte gebeurtenissen heel boos of juist heel verdrietig worden, omdat ze niet goed weten hoe ze hier mee om moeten gaan.
Ook hebben kinderen vaak voorkeur voor bepaald speelgoed of een bepaalde hobby waar ze zich heel lang mee kunnen vermaken.

AD(H)D
AD(H)D komt vaker voor bij kinderen met dit syndroom. Kinderen met ADHD hebben moeite om bij een taakje langere tijd de aandacht te houden. Ze spelen maar kort met een bepaald speelgoed en gaan dan weer naar een ander stukje speelgoed. Kinderen zijn snel afgeleid door een geluid of een beweging in de kamer. Op school hebben kinderen moeite langer tijd hun aandacht bij het schoolwerk te houden.
Kinderen kunnen moeite hebben met stil zitten en bewegen het liefst de hele dag. Kinderen hebben de neiging om eerst te doen en dan pas te denken of dit wel verstandig is, dit wordt impulsief gedrag genoemd.
Hoe wordt de diagnose KCNMA1-syndroom gesteld?
Verhaal en onderzoek
Aan de hand van het verhaal van een baby of een groter kind die last krijgt van epilepsieaanvallen en of bewegingsstoornis zal vaak gedacht worden aan de mogelijkheid van epilepsie als gevolg van een foutje in het DNA. Er bestaan echter veel andere verschillende aandoeningen die allemaal soortgelijke klachten kunnen geven. Er zal dus aanvullend onderzoek nodig zijn om de diagnose te stellen.
DNA onderzoek
Door middel van bloedonderzoek kan gekeken worden naar een foutje in het erfelijk materiaal van het KCNMA1-gen. Dit kan gericht gebeuren wanneer er aan gedacht wordt omdat de aandoening in de familie voorkomt, wat meestal niet het geval zal zijn.
Tegenwoordig wordt vaak een nieuwe genetische techniek ingezet (whole exome sequencing genoemd) waarbij in een keer een heleboel mogelijke foutjes in het DNA onderzocht kunnen worden. Op deze manier wordt meestal ontdekt dat er sprake is van een KCNMA1-syndroom.
MRI hersenen
Bij kinderen met epilepsie en/of een bewegingsstoornis wordt vaak een MRI van de hersenen gemaakt om te kijken of door middel van deze MRI-scan een verklaring gevonden kan worden waarom het kind last heeft van epilepsie. Deze MRI scan laat bij kinderen met het KCNMA1-syndroom meestal geen bijzonderheden zien. Bij kinderen met verlies van de functie van het BK-kanaal kunnen de kleinere hersenen kleiner zijn dan gebruikelijk.
Stofwisselingsonderzoek
Door middel van bloed- en urineonderzoek kan bij kinderen met epilepsie of een bewegingsstoornis gekeken worden of er sprake is van een stofwisselingsziekte als oorzaak van de epilepsie. Bij dit onderzoek worden bij kinderen met een KCNMA1-syndroom geen bijzonderheden gevonden.
EEG
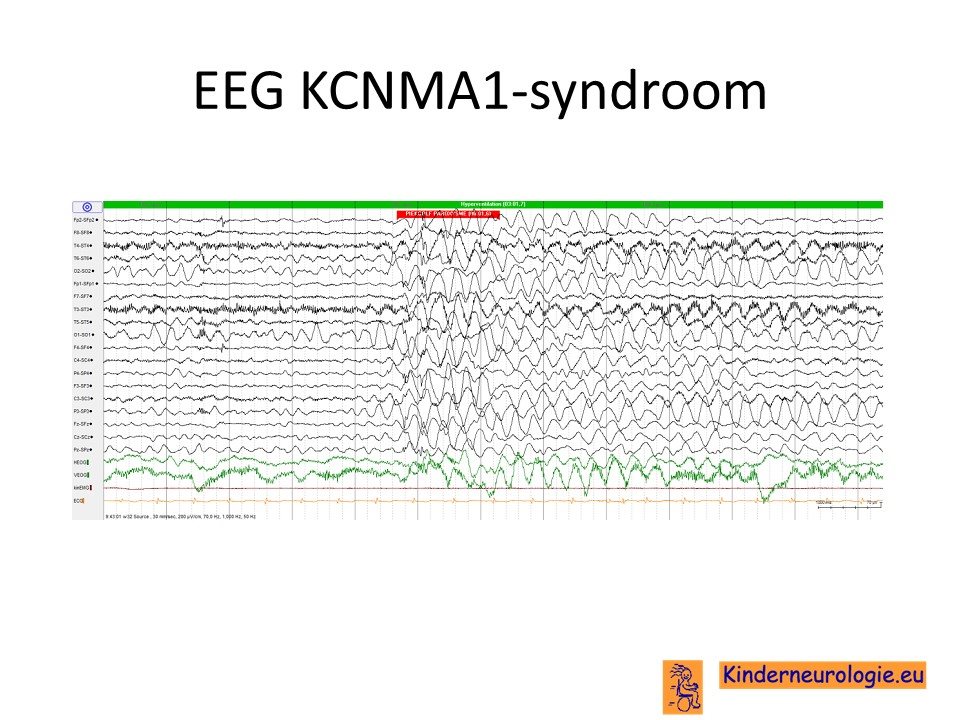
Op het EEG van kinderen met epilepsie als gevolg van het KCNMA1-syndroom worden vaak zogenaamde gegeneraliseerde epileptiforme afwijkingen gezien. Vaak gaat het om zogenaamde piekgolfcomplexen met een frequentie tussen de 3 en 4 Hz.
Hoe wordt KCNMA1-syndroom behandeld?
Geen genezing
Er bestaat geen behandeling die KCNMA1-syndroom kan genezen. De behandeling is er op gericht de epilepsie aanvallen en bewegingsstoornis zo veel mogelijk te onderdrukken, zodat kinderen energie krijgen om in hun ontwikkeling te steken.
Rust en regelmaat
Slaapgebrek en stress zijn in staat om aanvallen als gevolg van het hebben van het KCNMA1-syndroom uit te lokken. Daarom is een regelmatig dagritme met voldoende slaap belangrijk voor kinderen met KCNMA1-syndroom. Stress is niet altijd te voorkomen, maar bepaalde vormen van stress wel. Het is belangrijk om ook regelmatig rust en ontspanningsmomenten in de dag in te bouwen.
Aanvalsbehandeling epilepsie
De meeste epilepsieaanvallen gaan vanzelf over binnen enkele minuten. Omstanders hoeven dan niets te doen om de aanval te doen stoppen. Het is belangrijk om zo rustig mogelijk te blijven en het kind zo veel mogelijk met rust te laten.
Wanneer een aanval na 5 minuten nog niet vanzelf gestopt is, dan zal vaak geadviseerd worden om medicijnen te geven om een aanval te doen stoppen. De behandelende arts zal altijd aangeven welk tijdstip voor een bepaald kind het beste is. Medicijnen die gebruikt kunnen worden voor het stoppen van een aanval zijn diazepam rectiole (Stesolid®), midazolam neusspray, midazolam rectiole, lorazepam of clonazepam druppels.
Het effect van deze medicijnen ontstaat na enkele minuten. Nadien zal het kind meestal in slaap vallen, soms ook niet.

Behandeling epilepsie
Met behulp van medicijnen wordt geprobeerd om de epilepsieaanvallen zo veel mogelijk te voorkomen en het liefst er voor te zorgen dat er helemaal geen epilepsieaanvallen meer voorkomen. Soms lukt dit eenvoudig met een medicijn, maar het kan ook zijn dat een combinatie van medicijnen nodig is. Er bestaat geen duidelijk voorkeursmedicijn voor de behandeling van de epilepsie als gevolg van deze aandoening. Verschillende medicijnen kunnen gebruikt worden zoals fenobarbital, vigabatrine, prednison, clobazam, levetiracetam, zonisamide, acetozolamide of stiripentol.
Bij een deel van de kinderen zal het niet lukken om de epilepsieaanvallen met medicijnen onder controle te krijgen. Er bestaan ook andere behandelingen die een goed effect kunnen hebben op de epilepsie, zoals een ketogeen dieet, een nervus vagusstimulator, of een behandeling met methylprednisolon. Ook een combinatie van deze behandelingen met medicijnen die epilepsie onderdrukken is goed mogelijk

Behandeling bewegingsstoornis
De aanvallen van bewegingsstoornis kunnen verminderd worden door het gebruik van het medicijn clonazepam of diazepam. Ook andere medicijnen die gebruikt worden voor de behandeling van epilepsie, zoals levetiracetam, kunnen de aanvallen van de bewegingsstoornis verminderen. Een deel van de kinderen met drop attacks reageert op een behandeling met acetozolamide. Ook zijn er kinderen die reageren op lisdexamfetamine.
Fysiotherapie
Een fysiotherapeut kan adviezen geven aan de ouders hoe zij hun kind zo goed mogelijk kunnen helpen bij het stimuleren van de ontwikkeling.
Helm
Door de drop attacks kunnen kinderen op hun hoofd vallen en hoofdletsel oplopen. Een beschermhelm kan helpen om de kans op hersenletsel te verkleinen.

Onderzoek
Er wordt onderzoek gedaan naar medicijnen die het overactieve kaliumkanaal kunnen remmen. Er wordt gekeken of docosahexaeenzuur (DHA) een bepaald meervoudig onverzadigd vetzuur (omega-3-vetzuur) zou kunnen helpen bij deze aandoening. Dit vetzuur heeft invloed op de werking van kaliumkanalen. Een ander medicijn waar onderzoek naar wordt gedaan is naar vloeibaar albuterol. Ook dit medicijn heeft invloed op de werking van kaliumkanalen. Of dit daadwerkelijk gaat helpen bij kinderen met het KCNMA1-syndroom moet onderzocht worden. Daarnaast wordt gekeken of medicijnen die calciumblokkeren (zoals verapamil) van invloed kunnen zijn op het beloop van deze aandoening. Ook blijken medicijnen die gebruikt worden voor de behandeling van ADHD (dexamfetamine, lisdesamfetamine) bij sommige kinderen een effect te hebben op de drop attacks, dit wordt ook nader onderzocht.
Financiële kant van zorg voor een kind met een beperking
De zorg voor een kind met een beperking brengt vaak extra kosten met zich mee. Er bestaan verschillende wetten die zorg voor kinderen met een beperking vergoeden.
Daarnaast bestaan regelingen waar ouders een beroep op kunnen doen, om een tegemoetkoming te krijgen voor deze extra kosten. Meer informatie hierover vindt u in de folder financiën kind met een beperking.

Begeleiding
Een maatschappelijk werkende of psycholoog kan begeleiding geven hoe het hebben van deze aandoening een plaatsje kan krijgen in het dagelijks leven. Het kost vaak tijd voor ouders om te verwerken dat de toekomstverwachtingen van hun kind er anders uit zien dan mogelijk verwacht is. Ook vinden veel ouders het vaak lastig hoe zij hun tijd en aandacht moeten verdelen tussen het kind met de beperking en andere kinderen in het gezin. In de folder aandacht en tijd voor brussen vindt u tips die u hierbij kunnen helpen.

Door middel van een oproepje op het forum van deze site kunt u proberen in contact te komen met andere kinderen en hun ouders/verzorgers die ook te maken hebben met een KCNMA1-syndroom.
Wat betekent het hebben van een KCNMA1-syndroom voor de toekomst?
Weinig bekend
Omdat dit een aandoening is die nog niet lange tijd bekend is, is er weinig bekend over volwassenen met deze aandoening.
Transitie van zorg
Tussen de leeftijd van 16 en 18 jaar wordt de zorg vaak overgedragen van kinderspecialisten naar specialisten die de zorg aan volwassenen geven. Het is belangrijk om tijdig hierover na te denken. Is er behoefte de zorg over te dragen naar specialisten voor volwassenen of kan de huisarts de zorg leveren die nodig is.En als er behoefte is aan overdragen van de zorg naar specialisten voor volwassenen, naar welke dokter(s) wordt de zorg dan overgedragen? In welk ziekenhuis kan de zorg het beste geleverd worden. Het proces van overdragen van de zorg wordt transitie genoemd. Het is belangrijk hier tijdig over na te denken en een plan voor te maken samen met de dokters die betrokken zijn bij de zorg op de kinderleeftijd.
Ook verandert er veel in de zorg wanneer een jongere de leeftijd van 18 jaar bereikt. Voor meer informatie over deze veranderingen verwijzing wij u naar het artikel veranderingen in de zorg 18+.
![]()
Voorzichtig met alcohol
Alcohol is in staat om aanvallen als gevolg van het KCNMA1-syndroom uit te lokken. Het wordt daarom volwassenen met dit syndroom aangeraden om geen of anders zo min mogelijk alcohol te drinken.

Levensverwachting
Er is weinig bekend over de levensverwachting van kinderen met het KCNMA1-syndroom. Een moeilijk behandelbare vorm van epilepsie kan van invloed zijn op de levensverwachting. De oudste patient bekend met deze aandoening, is inmiddels ouder dan 50 jaar.
Kinderen krijgen
Wanneer een volwassene met de autosomaal dominante vorm van het KCNMA1-syndroom kinderen krijgt dan hebben deze kinderen 50% kans om zelf ook het KCNMA1-syndroom te krijgen. Of deze kinderen in dezelfde mate, in minder mate of in ernstigere mate last zullen krijgen van dit syndroom valt van te voren niet te voorspellen.
In geval van de autosomaal recessieve vorm van het KCNMA1-syndroom is de kans erg klein dat kinderen van een volwassene zelf ook dit syndroom krijgen. Dit kan alleen wanneer de partner drager is van een foutje in het KCNMA1-gen of zelf ook het KCNMA1-syndroom heeft. De kans hierop is klein.
Indien de volwassene geen kinderen wil of kan krijgen, kan nagedacht moeten worden over anticonceptie, waarover u in deze folder meer informatie vindt.
Hebben broertjes en zusjes ook een verhoogde kans om ook KCNMA1-syndroom te krijgen?
KCNMA1-syndroom wordt veroorzaakt door een fout in het erfelijke materiaal van het 10e-chromosoom. Vaak is het foutje bij het kind zelf ontstaan en niet overgeërfd van de vader of de moeder. Broertjes en zusjes hebben dan nauwelijks een verhoogde kans om zelf ook KCNMA1-syndroom te krijgen. Dit zou alleen kunnen wanneer de moeder het foutje in haar eicel of de vader het foutje in de zaadcel heeft zitten zonder dat dit in de andere lichaamscellen aanwezig is. De kans hierop is klein, ongeveer 1%. Dit wordt ouderlijk mocaisisme genoemd.
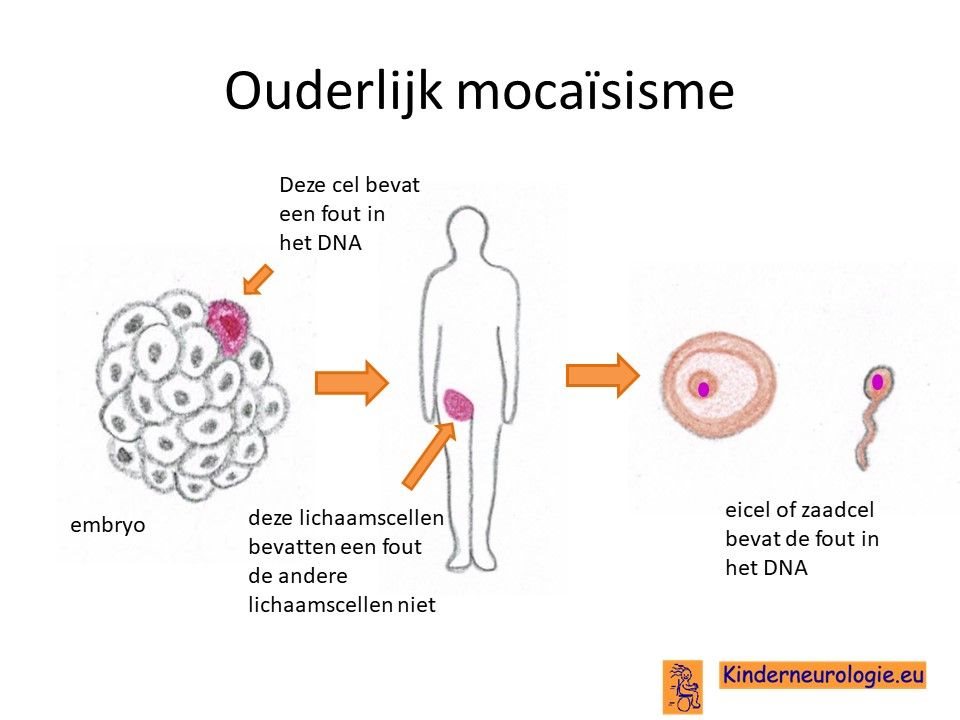
Wanneer een van de ouders zelf een foutje in het KCNMA1-gen heeft, dan hebben broertjes en zusjes 50% kans om zelf ook dit foutje te krijgen en een KCNMA1-syndroom te krijgen.
Een klinisch geneticus kan hier meer informatie over geven.
In geval van de zeldzamer voorkomende autosomaal recessieve vorm van het KCNMA1-syndroom, hebben broertjes en zusjes 25% kans om zelf ook het KCNMA1-syndroom te krijgen.
Een klinisch geneticus kan hier meer over vertellen.
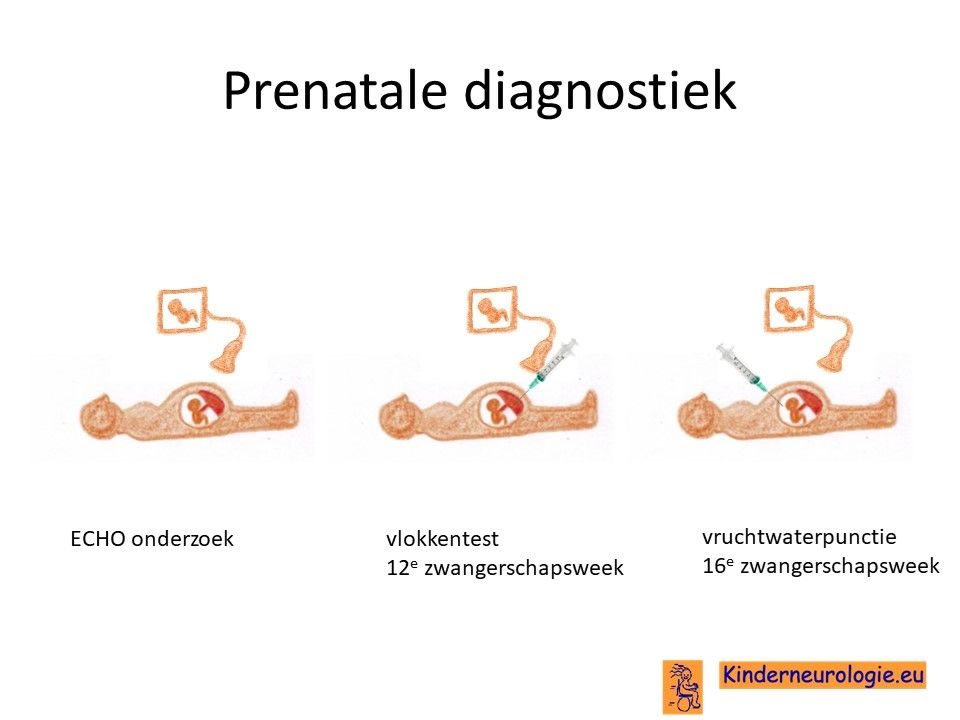
Prenatale diagnostiek
Wanneer bekend is welk foutje in een familie heeft gezorgd voor het ontstaan van KCNMA1-syndroom, dan is het mogelijk om tijdens een zwangerschap prenatale diagnostiek te verrichten in de vorm van een vlokkentest in de 12e zwangerschapsweek of een vruchtwaterpunctie in de 16e zwangerschapsweek. Beide ingrepen hebben een klein risico op het ontstaan van een miskraam (0,5% bij de vlokkentest en 0,3% bij de vruchtwaterpunctie). De uitslag van deze onderzoeken duurt twee weken. Voor prenatale diagnostiek kan een zwangere de 8ste week verwezen worden door de huisarts of verloskundige naar een afdeling klinische genetica. Meer informatie over prenatale diagnostiek kunt u vinden op de website: www.pns.nl
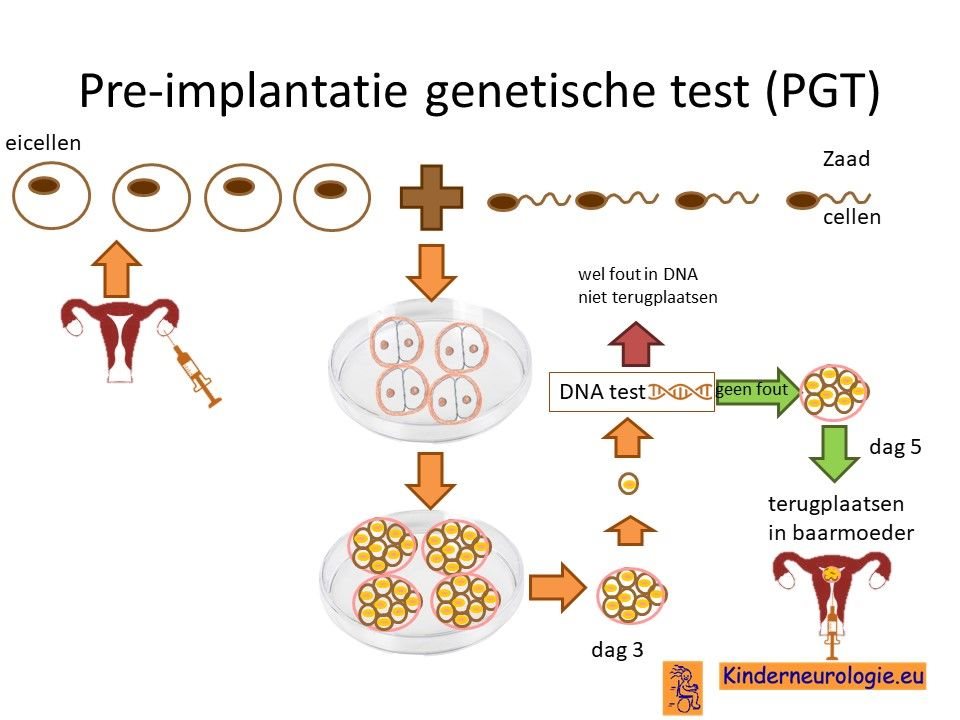
Preïmplantatie Genetische Test (PGT)
Stellen die eerder een kindje hebben gehad met een ernstige vorm van het KCNMA1-syndroom kunnen naast prenatale diagnostiek ook in aanmerking voor een preïmplantatie genetische test (PGT.) Bij PGT wordt een vrouw zwanger door middel van IVF (In Vitro Fertilisatie). De bevruchting vindt dan buiten het lichaam plaats, waardoor het zo ontstane pre-embryo onderzocht kan worden op het hebben van KCNMA1-syndroom. Alleen embryo’s zonder de aanleg voor KCNMA1-syndroom, komen in aanmerking voor terugplaatsing in de baarmoeder. Voor meer informatie zie www.pgtnederland.nl.
Wilt u dit document printen dan kunt u hiervoor de printbutton bovenaan de pagina gebruiken
Wilt u ook uw verhaal kwijt, dat kan: verhalen kunnen gemaild worden via via contact en zullen daarna zo spoedig mogelijk op de site worden geplaatst. Voor meer informatie zie hier.
Heeft u foto's die bepaalde kenmerken van deze aandoening duidelijk maken en die hier op de website mogen worden geplaatst, dan vernemen wij dit graag via contact.
Links
www.epilepsievereniging.nl
(site van epilepsievereniging Nederland)
www.epilepsie.nl
(site van het nationaal epilepsiefonds)
www.kciaf.org/
(Engelstalige site over KCNMA1-syndroom)
Referenties
- De novo KCNMA1 mutations in children with early-onset paroxysmal dyskinesia and developmental delay. Zhang ZB, Tian MQ, Gao K, Jiang YW, Wu Y. Mov Disord. 2015;30:1290-2.
- Homozygous KCNMA1 mutation as a cause of cerebellar atrophy, developmental delay and seizures. Tabarki B, AlMajhad N, AlHashem A, Shaheen R, Alkuraya FS. Hum Genet. 2016;135:1295-1298
- Paroxysmal dyskinesias revisited: a review of 500 genetically proven cases and a new classification. Erro R, Sheerin UM, Bhatia KP. Mov Disord. 2014;29:1108-16.
- Drop Attacks in patients with KCNMA1 p.N999S heterozygous de novo mutations Heim J, Vermuri A, Meredith A, Keros S, Kruer M Oral presentation 6th International symposium on paediatric movement disorders Barcelona 2019
- De novo loss-of-function KCNMA1 variants are associated with a new multiple malformation syndrome and a broad spectrum of developmental and neurological phenotypes. Liang L, Li X, Moutton S, Schrier Vergano SA, Cogné B, Saint-Martin A, Hurst ACE, Hu Y, Bodamer O, Thevenon J, Hung CY, Isidor B, Gerard B, Rega A, Nambot S, Lehalle D, Duffourd Y, Thauvin-Robinet C, Faivre L, Bézieau S, Dure LS, Helbling DC, Bick D, Xu C, Chen Q, Mancini GMS, Vitobello A, Wang QK. Hum Mol Genet. 2019;28:2937-2951
- KCNMA1-linked channelopathy. Bailey CS, Moldenhauer HJ, Park SM, Keros S, Meredith AL. J Gen Physiol. 2019;151:1173-1189
- Two novel presentations of KCNMA1-related pathologyExpanding the clinical phenotype of a rare channelopathy. Rodrigues Bento J, Feben C, Kempers M, van Rij M, Woiski M, Devriendt K, De Catte L, Baldewijns M, Alaerts M, Meester J, Verstraeten A, Hendson W, Loeys B. Mol Genet Genomic Med. 2021;9:e1797
Laatst bijgewerkt: 27 maart 2022 voorheen: 25 novemner 2020, 8 maart 2020 voorheen: 6 mei 2019 voorheen: 16 maart 2019 en 18 oktober 2018
Auteur: JH Schieving