Wat is het Smith-Kingsmore syndroom?
Het Smith-Kingsmore syndroom is een erfelijke aangeboren aandoening waarbij kinderen en volwassenen een combinatie aan problemen kunnen krijgen zoals een groot hoofd en een grote lengte, typische uiterlijke kenmerken, een lage spierspanning vaak in combinatie met problemen met leren.
Hoe wordt het Smith-Kingsmore syndroom ook wel genoemd?
Het Smith-Kingsmore syndroom is genoemd naar twee artsen die dit syndroom beschreven hebben in 2013. Het wordt ook wel afgekort met de letters SKS.
MINDS- syndroom
Een andere naam die ook gebruikt wordt is het MINDS-syndroom. De M staat voor macrocefalie wat groot hoofd betekent. De I staat voor intellectual disability wat aangeeft dat kinderen met dit syndroom problemen hebben met leren. De N en de D staan voor neurodevelopmental disorder en geven aan dat de ontwikkeling van kinderen met deze aandoening anders verloopt dan die van andere kinderen. De S staat voor small thorax en geeft aan dat een deel van deze kinderen een smalle borstkas heeft. Anderen vinden dat de S staat voor syndroom.
Overgrowth syndroom
Het Smith-Kingsmore syndroom behoort tot de groep van overgrowth syndromen. Dit zijn aandoeningen waarbij kinderen een grote lengte en een groot hoofd hebben. Andere overgrowth syndromen zijn het Sotos syndroom, het Cowden syndroom, het MPPH-syndroom en het Waever syndroom.
Hoe vaak komt Smith-Kingsmore syndroom voor?
Smith-Kingsmore syndroom is een zeldzame ziekte, het is nog niet bekend hoe vaak deze aandoening voorkomt bij kinderen en volwassenen. Omdat het Smith-Kingsmore syndroom relatief onbekend is, zal de diagnose bij een deel van de mensen die dit syndroom wel hebben nog niet gesteld zijn. Dankzij nieuwe genetische technieken zal het steeds gemakkelijker worden om deze diagnose te stellen. Dan zal ook pas duidelijk worden hoe vaak deze aandoening voorkomt. Anno 2021 zijn er iets meer dan 200 kinderen wereldwijd bekend met deze aandoening.

Bij wie komt het Smith-Kingsmore syndroom voor?
Het Smith-Kingsmore syndroom is al voor de geboorte aanwezig. Het grotere hoofd en de lagere spierspanning, vallen al op tijdens het eerste levensjaar.
Het Smith-Kingsmore syndroom komt even vaak bij jongens als bij meisjes voor.
Wat is de oorzaak van het Smith-Kingsmore syndroom?
Fout in erfelijk materiaal
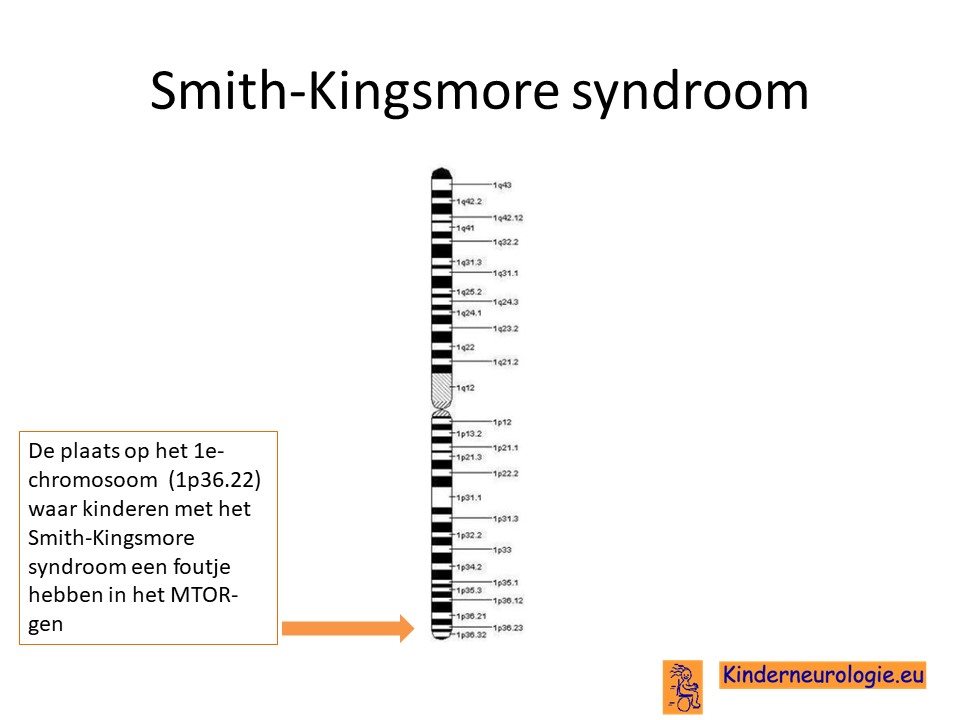
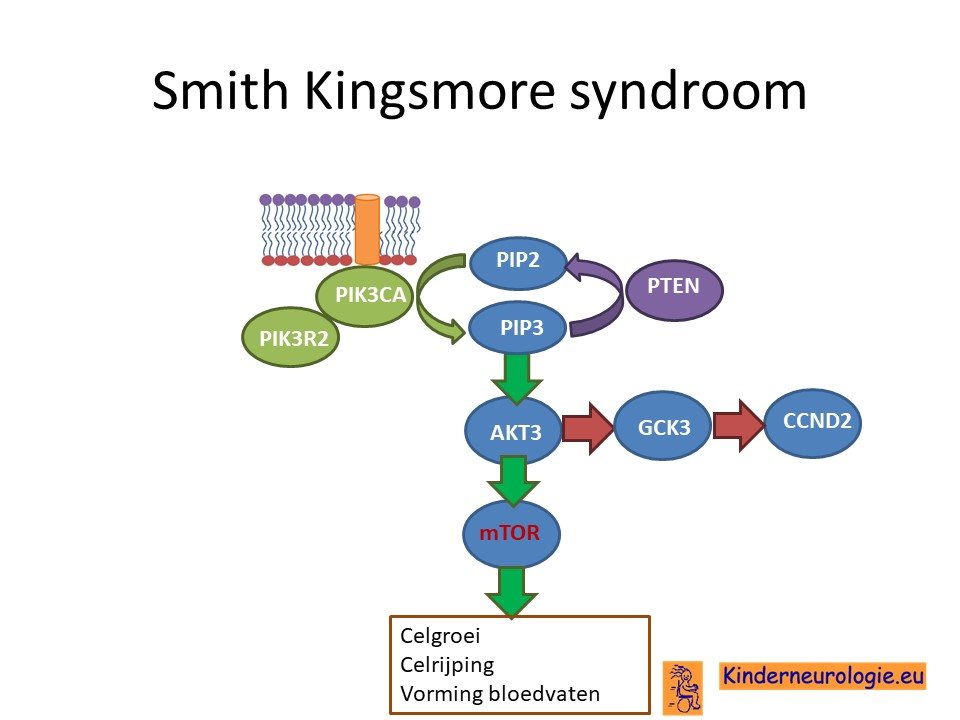
Kinderen met het Smith-Kingsmore syndroom hebben een fout in het erfelijk materiaal van chromosoom 1. De plaats van deze fout wordt het MTOR-gen genoemd.
Autosomaal dominant
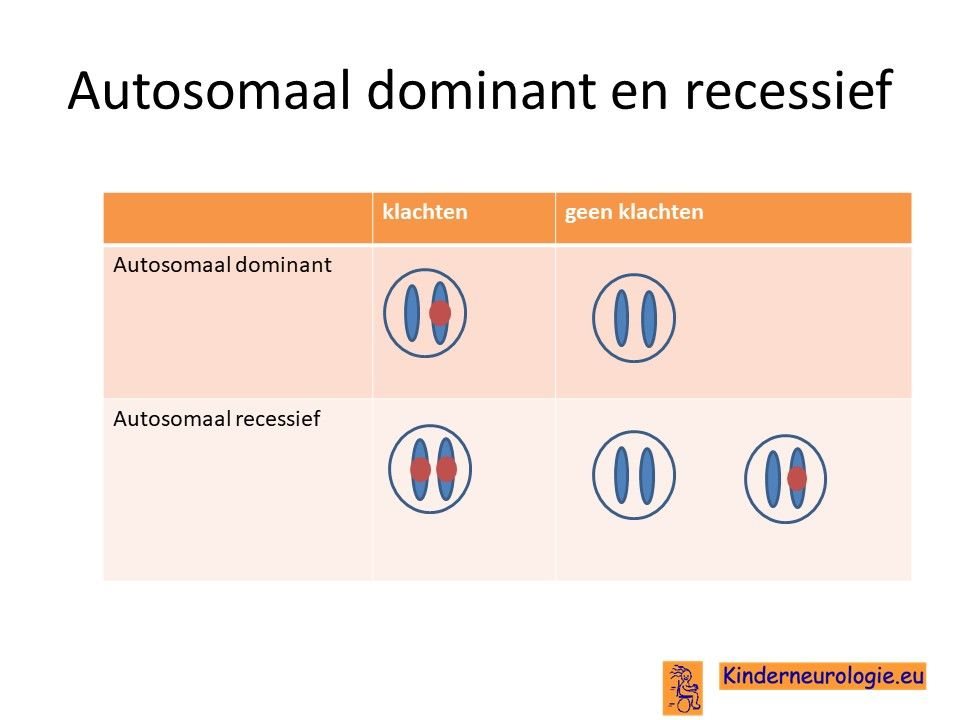
De fout in het DNA is een zogenaamd autosomaal dominant fout Dat wil zeggen dat een foutje op een van de twee chromosomen 1 die een kind heeft in het MTOR-gen al voldoende is om de aandoening te krijgen. Dit in tegenstelling tot een autosomaal recessief fout waarbij kinderen pas klachten krijgen wanneer beide chromosomen een fout bevatten.
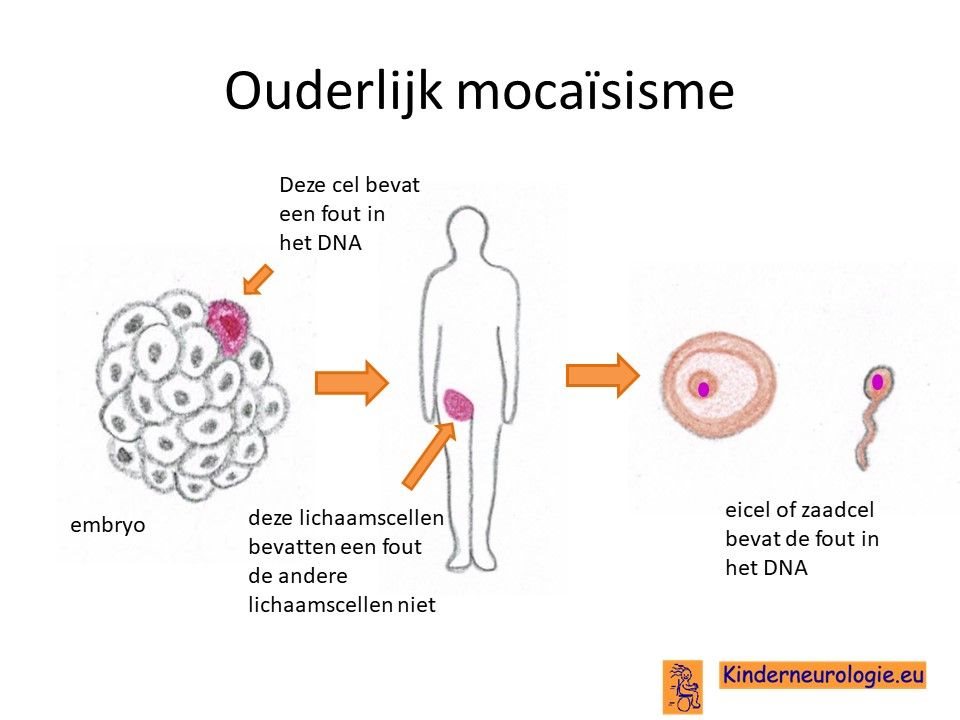
Bij het kind zelf ontstaan
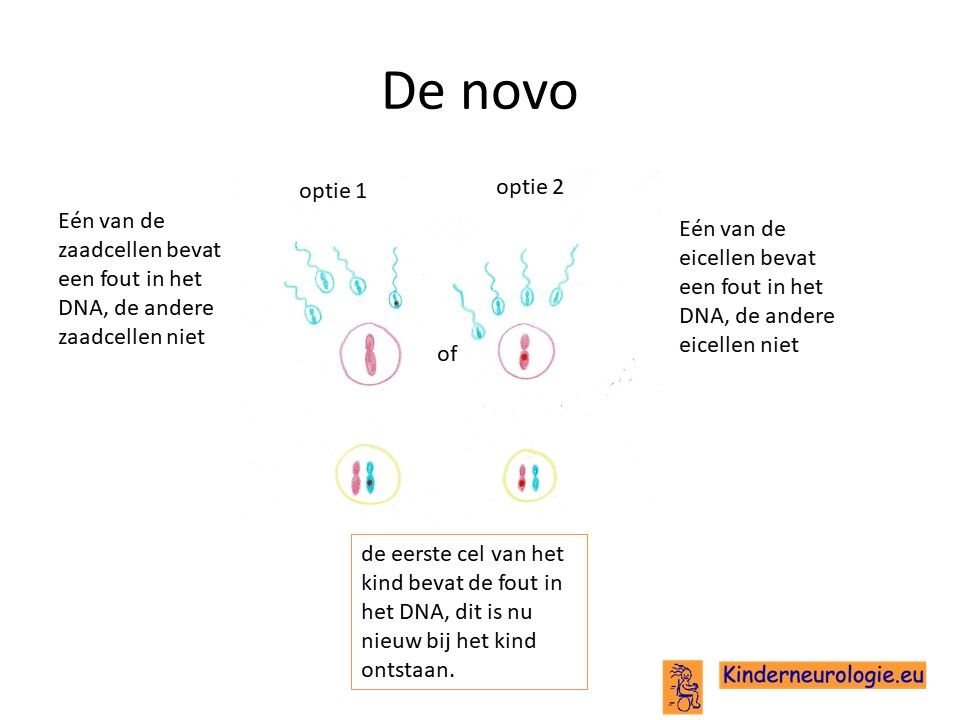
Bij een deel van de kinderen met het Smith-Kingsmore syndroom is de fout bij het kind zelf ontstaan tijdens de bevruchting van de eicel door de zaadcel.
Geërfd van een ouder
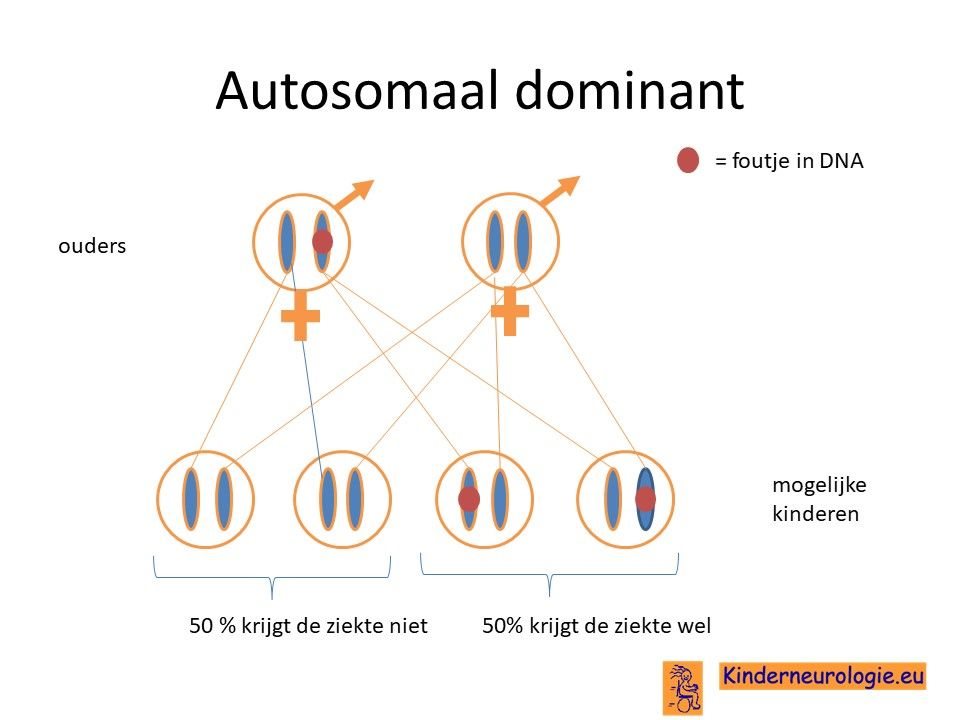
Een deel van de kinderen heeft de fout in het MTOR-gen geërfd van een ouder die zelf ook deze fout in het MTOR-gen heeft Soms was al bekend dat de ouder zelf ook het Smith-Kingsmore syndroom heeft, soms wordt de diagnose bij de ouder pas gesteld wanneer de aandoening bij het kind wordt gesteld.
Afwijkend eiwit
Als gevolg van de fout in het MTOR-gen wordt een bepaald eiwit niet goed aangemaakt. Dit eiwit heet mechanistic target of rapamycin, ook wel afgekort als MTOR eiwit. Het MTOR-eiwit is een heel belangrijk eiwit in het lichaam. Het regelt allerlei belangrijke processen in het lichaam. Zo is het MTOR eiwit belangrijk bij toename van het aantal lichaamscellen voor en na de geboorte. Door de fout in het MTOR-gen krijgen kinderen met dit syndroom meer lichaamscellen waardoor ze een grote lengte krijgen. Ook zorgt het MTOR eiwit voor de aanmaak van stukjes mRNA die nodig zijn voor de aanmaak van allerlei belangrijke eiwitten. Het mTOR eiwit speelt ook een belangrijke rol bij de aanmaak van eiwitten die zorgen voor stevigheid van cellen.
Aanleg van de hersenen
Het MTOR eiwit speelt een belangrijke rol bij de aanleg van de hersenen. Door de fout in het MTOR eiwit krijgen kinderen met het Smith-Kingsmore syndroom meer hersencellen dan gebruikelijk. Hierdoor krijgen kinderen een groter hoofd. Dit te veel aan hersencellen zit de werking van de hersenen in de weg, de hersenen werken hierdoor juist langzamer dan hersenen met het normale aantal hersencellen. Ook speelt het MTOR eiwit een belangrijke rol bij de uitrijping van de hersencellen. De hersenen van kinderen met dit syndroom zijn ook vaak anders aangelegd dan de hersenen van kinderen zonder dit syndroom. Aanlegstoornissen zoals een corticale dysplasie komen vaak voor bij kinderen met dit syndroom.
Wat zijn de symptomen van Smith-Kingsmore syndroom?
Grote variatie
De symptomen die voorkomen bij Smith-Kingsmore syndroom kunnen sterk uiteenlopen. Sommige kinderen en volwassenen hebben alle verschijnselen, andere maar enkele. Het valt vooraf niet te voorspellen hoeveel en welke symptomen kinderen zullen krijgen.

Jouw kind is uniek
Bedenk dat onderstaande symptomen kunnen voorkomen bij jouw kind, maar ook niet allemaal zullen voorkomen. Jouw kind is uniek en veel meer dan een kind met deze aandoening. Het lezen van mogelijke symptomen die kunnen voorkomen, kan ouders het gevoel geven dat er alleen maar aandacht is voor de beperkingen van het kind. Dat is zeer zeker niet de bedoeling. Jouw kind is bijvoorbeeld lief, grappig, gevoelig, gezellig,sociaal, vindingrijk, nieuwsgierig, ondeugend, enthousiast,een zonnestraaltje, creatief en/of innemend en dat vind je niet terug in onderstaande symptomen die kunnen horen bij dit syndroom. Dat kan ook niet, want die eigenschappen maken jouw kind nu eenmaal uniek. Blijf daar vooral naar kijken en zie deze symptomen meer als achtergrondinformatie die je kunnen helpen om te begrijpen wat er met je kind aan de hand zou kunnen zijn wanneer jouw kind zich anders ontwikkelt of ergens last van heeft. Deze informatie kan jullie als ouders en hulpverleners een handvat geven wat hiervoor een mogelijke verklaring kan zijn.

Hoog geboortegewicht
De meeste baby’s worden geboren met een hoger geboortegewicht dan gebruikelijk. Voldagen baby’s wegen vaak meer dan 4 kilo. Een hoog geboorte gewicht wordt ook wel macrosomie genoemd.
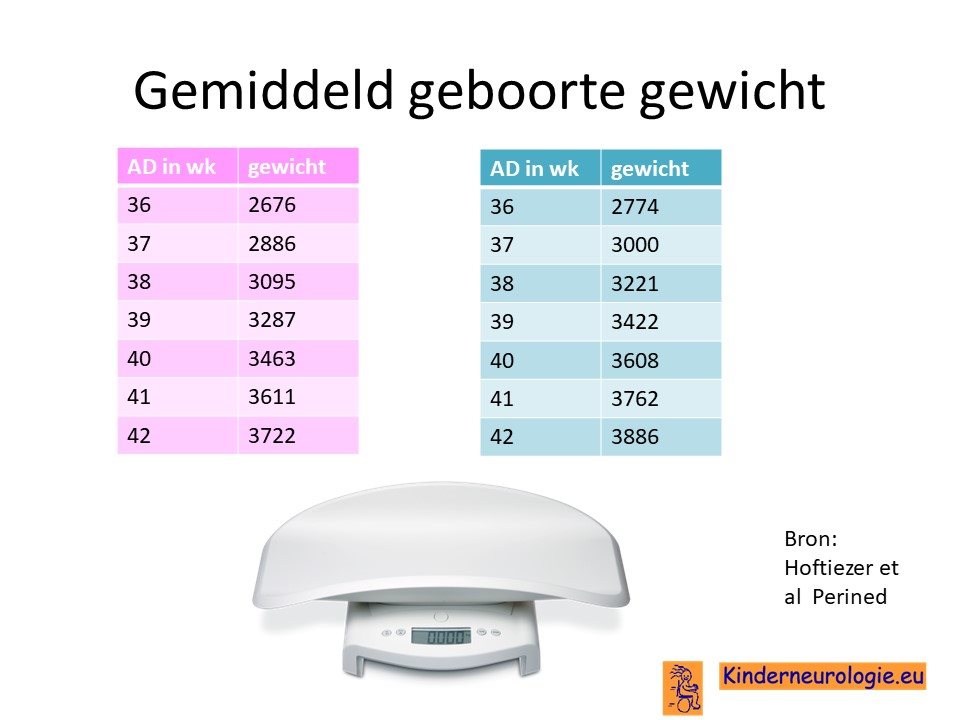
Grote lengte
Ook zijn baby’s met dit syndroom bij de geboorte al langer dan gebruikelijk.
Groot hoofd
Een deel van de baby’s heeft bij de geboorte al een grotere hoofdomtrek. Dit kan zorgen voor problemen bij de bevalling, het hoofdje kan hierdoor moeizamer geboren worden. Tijdens het eerste levensjaar groeit het hoofd van kinderen met dit syndroom sneller dan van andere kinderen. De grootte van het hoofd komt dan buiten de lijntjes van de groeicurve te lopen. Vaak wordt er gedacht dat er misschien sprake is van een waterhoofd, maar dit is niet het geval bij kinderen met dit syndroom. Een groot hoofd is zwaar. Het kost kinderen met dit syndroom daarom mee moeite en tijd om te leren hun hoofd op te tillen. Dit heeft gevolgen voor het leren zitten, kruipen en staan. Ook kan het lastig zijn om kleding te vinden die goed over het hoofd heen gaat. Met het ouder worden, valt het grotere hoofd minder op, omdat kinderen zelf ook lang zijn. Wel kan het lastig blijven om bijvoorbeeld een cap voor paardrijden of een helm voor op de fiets te vinden.
Grote fontanel
Baby’s hebben ruimte tussen de botten van de schedel. Boven op het hoofd zit een grote ruimte. Dit wordt de grote fontanel genoemd. Bij kinderen met dit syndroom is deze open ruimte vaak extra groot.
Problemen met drinken
Baby’s met dit syndroom hebben vaak problemen met drinken. Ze pakken de borst of speen niet goed, drinken onregelmatig en stoppen snel met drinken. Het kost vaak veel tijd om baby’s met dit syndroom de borst of de fles te geven, maar uiteindelijk lukt dit wel. Zelden is het nodig om kinderen tijdelijk sondevoeding te geven omdat zij anders niet genoeg voeding binnen krijgen. Met het ouder worden, verloopt het drinken wel beter.
Lage bloedsuiker
Baby’s met dit syndroom krijgen gemakkelijker last van een te lage bloedsuiker waarde. Een te lage bloedsuiker waarde kan ontstaan wanneer kinderen langere tijd niet drinken. Door de lage bloedsuiker waarde kunnen kinderen trillerig worden.
Lagere spierspanning
Kinderen met het Smith-Kingsmore syndroom hebben vaak een lagere spierspanning. De armen en benen voelen wat slapper aan, gewrichten kunnen gemakkelijker overstrekt worden. Kinderen zullen meer spierkracht nodig hebben om deze gewrichten stabiel te houden. Daarom gaat een groot deel van de kinderen met het Smith-Kingsmore syndroom ook later kruipen, staan en lopen dan andere kinderen. Op grotere leeftijd hebben kinderen vaak platvoeten de onderkant van de voet is niet hol, maar plat. Door de lagere spierspanning kunnen kinderen gemakkelijker hun enkels verzwikken.
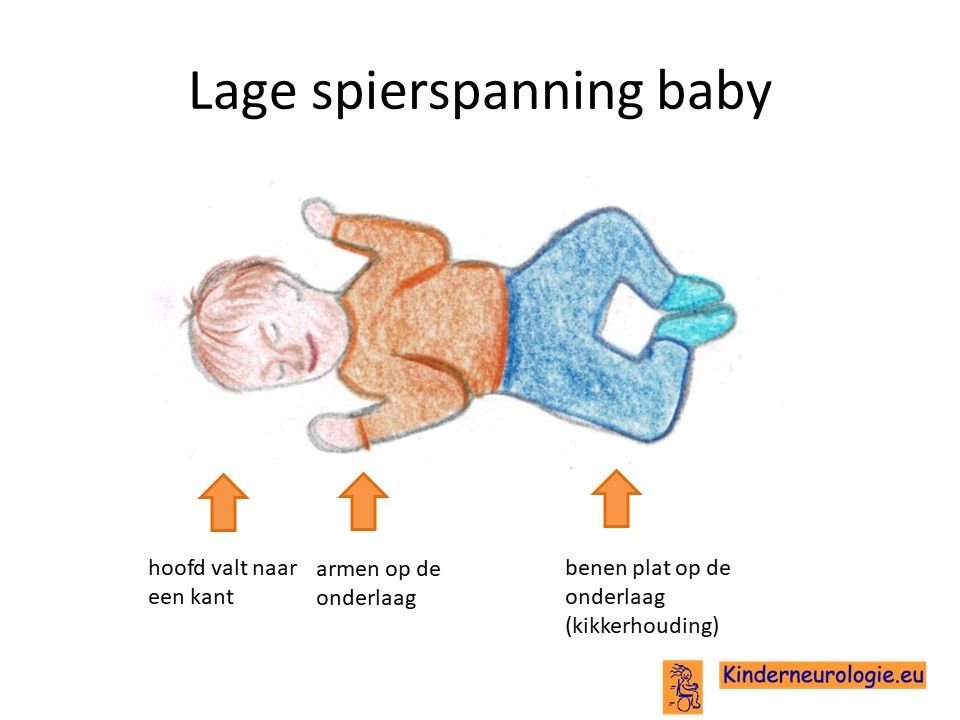
Ontwikkelingsachterstand
Door de combinatie van het grotere hoofd, de lagere spierspanning en de hersencellen die langzamer werken dan gebruikelijk ontwikkelen kinderen met het Smith-Kingsmore syndroom zich langzamer dan andere kinderen. Kinderen gaan later zitten, kruipen en lopen dan andere kinderen maar leren dit allemaal wel. Ook het leren van fietsen en zwemmen is voor kinderen met het Smith-Kingsmore syndroom lastiger dan voor andere kinderen. Het bewegen gaat vaak wat houteriger en minder soepel. Kinderen vallen gemakkelijker dan andere kinderen.
Veel kinderen hebben ook meer moeite met het leren van tekenen, schrijven en knippen. Dit kost voor hen ook meer tijd om dit te leren. Dit worden fijn motorische problemen genoemd.
Spraaktaalontwikkeling
Bij een deel van de kinderen komen ook de eerste woordjes en zinnetjes later dan bij kinderen zonder dit syndroom, bij andere kinderen is dit niet zo. De meeste kinderen zijn in staat om in zinnen te praten. Wel kunnen kinderen het lastig vinden om te vertellen wat ze hebben meegemaakt of om aan te geven hoe ze zich voelen. Omdat de spieren van de mond vaak slapper zijn dan gebruikelijk, zijn sommige kinderen lastiger te verstaan dan andere kinderen. Voor vier op de tien kinderen is het te moeilijk om te leren praten, zij communiceren door middel van gezichtsuitdrukking en/of gebaren.
Problemen met leren
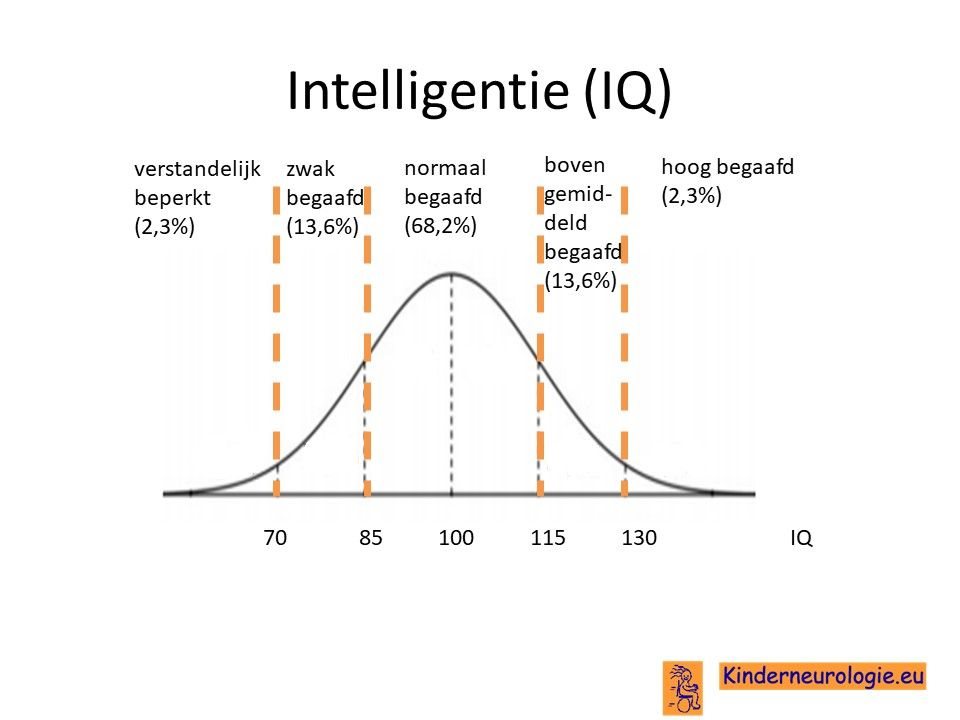
Kinderen met het Smith-Kingsmore syndroom hebben vaker problemen met leren. Kinderen hebben meer tijd en meer herhaling nodig om nieuwe stof aan te leren. De mate van problemen met leren kan erg verschillen van mild tot ernstig. Bij een deel van de kinderen is het IQ lager dan 70, de grens waaronder wordt gesproken van een verstandelijke beperking. Een op de vijf kinderen is er sprake van een IQ tussen de 70 en 85, bij hen wordt er gesproken van zwakbegaafdheid.
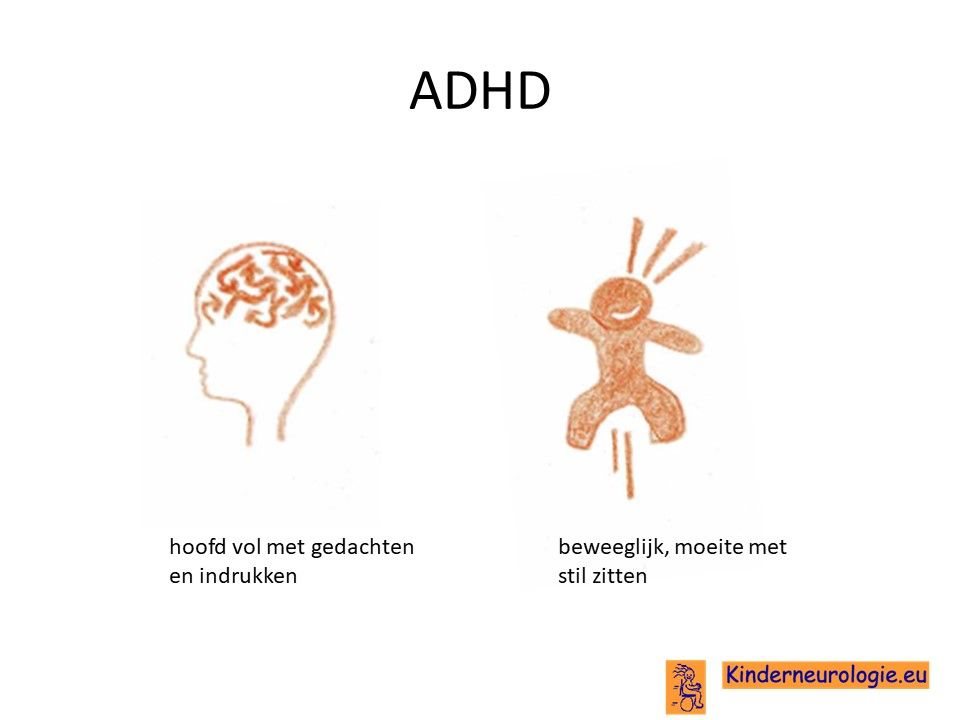
AD(H)D
AD(H)D komt vaker voor bij kinderen met dit syndroom. Kinderen met AD(H)D hebben moeite om bij een taakje langere tijd de aandacht te houden. Ze spelen maar kort met een bepaald speelgoed en gaan dan weer naar een ander stukje speelgoed. Kinderen zijn snel afgeleid door een geluid of een beweging in de kamer. Op school hebben kinderen moeite langer tijd hun aandacht bij het schoolwerk te houden.
Een deel van de kinderen heeft moeite met stil zitten en beweegt het liefst de hele dag. Dan wordt er gesproken van ADHD. Wanneer dit niet het geval is wordt gesproken van ADD.
Sociaal-emotionele ontwikkeling
Kinderen met Smith-Kingsmore syndroom zijn vaak rustige, tevreden baby’s die zich zelf goed kunnen vermaken. Ook wanneer kinderen ouder worden zijn ze vaak tevreden kinderen die zichzelf heel goed kunnen vermaken.
Stereotypieën
Veel kinderen met dit syndroom maken graag bewegingen met hun armen en hun handen die vaak terug keren. Zulke bewegingen worden stereotypieën genoemd. Sommige kinderen gaan wapperen met hun handen, anderen maken draaiende bewegingen of wrijvende bewegingen over de borst heen. Deze bewegingen komen vaak voor wanneer kinderen iets heel leuks of iets spannends gaan doen. Kinderen hebben hier zelf geen last van. Soms worden stereotypieen ook wel stimming genoemd.

Zelfverwondend gedrag
Een deel van de kinderen verwondt zich zelf door te bijten in de vingers, in de huid of door te krabben of aan haren te trekken. Deze vorm van zelfverwonding komt vaak voor wanneer kinderen gespannen of bang zijn of zichzelf vervelen.

Autistiforme kenmerken
Een deel van de kinderen met Smith-Kingsmore syndroom heeft autistiforme kenmerken (geschat vier op de tien kinderen) . Kinderen vinden het dan bijvoorbeeld moeilijk om oogcontact te maken of om contact te maken met leeftijdsgenootjes. Contact maken met volwassenen gaat vaak wel goed.
Sommige kinderen houden van een vast dagschema en vinden het heel moeilijk om te gaan met onverwachte gebeurtenissen hierin. Deze onverwachte gebeurtenissen kunnen kinderen heel boos of heel angstig maken. Zij hebben dan vaak de hulp van een volwassene nodig om hier weer uit te komen.

Angst
Kinderen met dit syndroom hebben gemakkelijker last van angsten. Bijvoorbeeld angst om alleen zonder de ouders te zijn, angst voor het donker of angst voor onbekende en vreemde situaties.
Epilepsie
Een deel van de kinderen met Smith-Kingsmore syndroom heeft epilepsie aanvallen. Verschillende type aanvallen kunnen voorkomen, aanvallen met schokken, of aanvallen met verstijven van een arm of been. Vier op de tien kinderen heeft een epilepsie aanval gehad, in die situatie wordt nog niet gesproken van epilepsie. De diagnose epilepsie wordt past gesteld wanneer er meerdere epilepsieaanvallen zijn geweest binnen een jaar.
Problemen met slapen
Slaapproblemen komen vaak voor bij kinderen met dit syndroom. Sommige kinderen hebben moeite met het inslapen. Een groot deel van de kinderen wordt ’s nachts regelmatig wakker en komt dan maar moeilijk weer in slaap. Sommige kinderen zijn vroeg in de ochtend wakker en zijn dan al meteen actief en willen niet meer slapen.
Bij een deel van de kinderen worden deze slaapproblemen veroorzaakt door epilepsie gedurende de nacht.

Uiterlijke kenmerken
Bij veel syndromen hebben kinderen vaak wat veranderde uiterlijke kenmerken. Hier hebben kinderen zelf geen last van, maar het kan de dokters helpen om te herkennen dat er sprake is van een syndroom en mogelijk ook van welk syndroom. Ook maakt dit vaak dat kinderen met hetzelfde syndroom vaak meer op elkaar lijken dan op hun eigen broertjes en zusjes, terwijl de kinderen toch niet familie van elkaar zijn.
Kinderen met het Smith-Kingsmore syndroom hebben een groot hoofd. Het voorhoofd is vaak hoog en kan wat naar voren toe bollen. De ogen staan vaak wat verder uit elkaar dan gebruikelijk en lopen in de richting van de oren een stukje naar beneden. De neusbrug is vaak diep en de neus zelf kort. De huid tussen de neus en de mond is vaak glad, het groefje wat veel kinderen hebben in dit stukje huid ontbreekt. Ook kan de afstand tussen de neus en de mond vergroot zijn. De mond is vaak breed, de bovenlip dun. Het gehemelte kan hoog zijn.
De kin is vaak smal.
Haren
Krullend of golvend haar komt vaker voor bij kinderen met dit syndroom dan bij kinderen zonder dit syndroom.
Lengte
Kinderen met het Smith-Kingsmore syndroom groeien vaak sneller dan hun leeftijdsgenoten. Zij zijn vaak langer dan hun leeftijdsgenoten. Dit kan maken dat kinderen overschat worden in hun leeftijd.
Handen en voeten
Kinderen met dit syndroom hebben vaak korte vingers en tenen. De nagels zijn vaak smal en breken gemakkelijker af. Ook kunnen de lijnen die in de hand en voet lopen diep zijn en daardoor extra opvallen. Soms zijn de handen of de onderarmen in verhouding kleiner dan verwacht mag worden op grond van de lengte.
Gewicht
Vanaf de puberteit hebben kinderen met het Smith-Kingsmore syndroom de neiging om zwaarder te worden dan hun leeftijdsgenoten. Zij komen gemakkelijker aan in gewicht. Dit extra gewicht raken ze niet zo maar meer kwijt. Kinderen en volwassenen met het Smith-Kingsmore syndroom hebben een verhoogde kans om overgewicht te krijgen.

Zien
Scheelzien komt vaker voor bij kinderen met Smith-Kingsmore syndroom. Door dit scheelzien bestaat er een verhoogde kans op het krijgen van een lui oog. Dit is een oog waar kinderen minder goed mee kunnen zien.
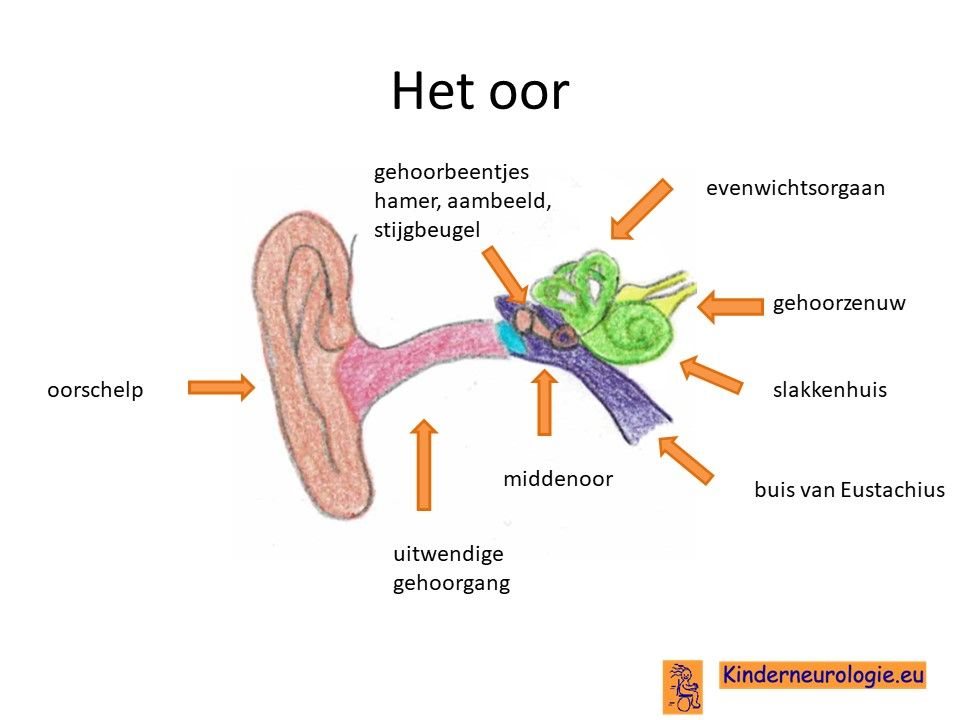
Oren
Kinderen met dit syndroom zijn gevoeliger voor het krijgen van middenoorontstekingen.
Open mond
Kinderen met dit syndroom hebben de neiging om door hun mond te ademen in plaats van door hun neus. Dit komt door zwakte van de spieren in het gezicht waardoor kinderen meer moeite moeten doen om hun mond dicht te houden. Wanneer ze hier niet aan denken, dan valt hun mond open.

Toegenomen eetlust
Kinderen met dit syndroom hebben vaak een grote eetlust. Zij voelen niet goed dat ze vol zitten en blijven maar eten.
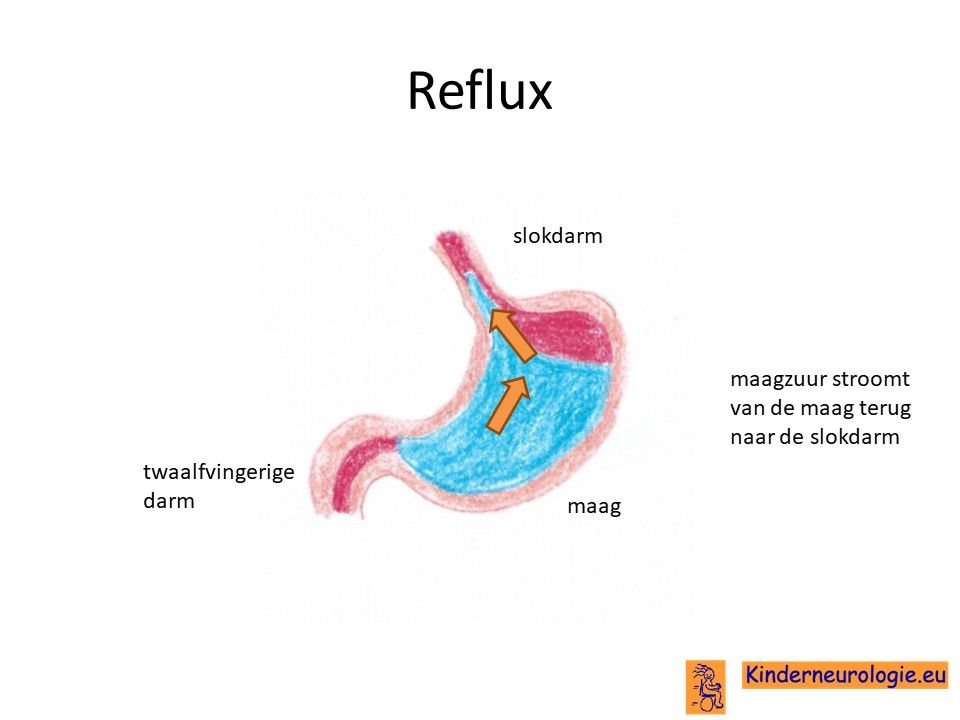
Reflux
Kinderen met dit syndroom hebben heel vaak last van het terugstromen van voeding vanuit de maag naar de slokdarm. Dit wordt reflux genoemd. Omdat de maaginhoud zuur is, komt het zuur zo ook in de slokdarm, soms zelfs ook in de mond. Dit zuur kan zorgen voor pijnklachten, waardoor kinderen moeten huilen en soms ook niet willen eten. Ook kan het maken dat kinderen moeten spugen. Door het zuur kan de slokdarm geïrriteerd en ontstoken raken. Wanneer dit niet tijdige ontdekt en behandeld wordt, kan dit zorgen voor het spuug met daarin bloedsliertjes.
Kwijlen
Kinderen met dit syndroom hebben gemakkelijk last van kwijlen. Dit komt door slapheid van de spieren in het gezicht en rondom de mond, waardoor het speeksel gemakkelijk uit de mond loopt.
Problemen met kauwen
Kinderen met dit syndroom hebben vaker een probleem met kauwen. Kinderen hebben het liefst zacht eten waarop ze niet hoeven te kauwen of eten wat in kleine stukjes gesneden is. Kauwen kost kinderen veel tijd. Taai vlees krijgen kinderen met deze aandoening maar moeilijk fijngemalen waardoor kinderen dit liever uitspugen dan doorslikken.

Borstkas
Een deel van de kinderen met dit syndroom heeft een smalle borstkas. Hier hebben kinderen zelf geen last van.
Aangeboren hartafwijking
Een klein deel van de kinderen heeft een aangeboren hartafwijking. Er bestaat dan een verbinding tussen de grote lichaamsslagader (aorta) en de rechterboezem.
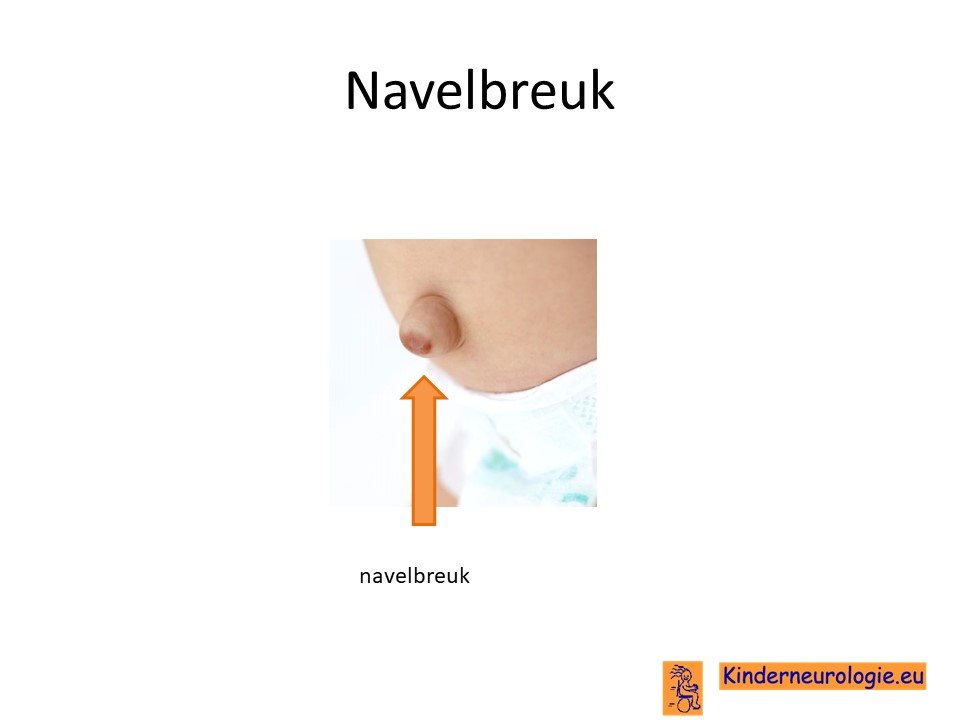
Navelbreuk
Een groot deel van de kinderen met dit syndroom heeft een navelbreuk. De navel staat dan bol. Soms is dit weinig opvallend, maar soms is er ook sprake van een grote zwelling op de buik die groter wordt bij persen. De rechte buikspieren liggen vaak niet goed tegen elkaar aan, waardoor er ruimte zit tussen deze rechter buikspieren.
Aangeboren nierafwijking
Bij een klein deel van de kinderen zijn beide nieren niet even groot. Hier hoeven kinderen geen last van te hebben of te krijgen.
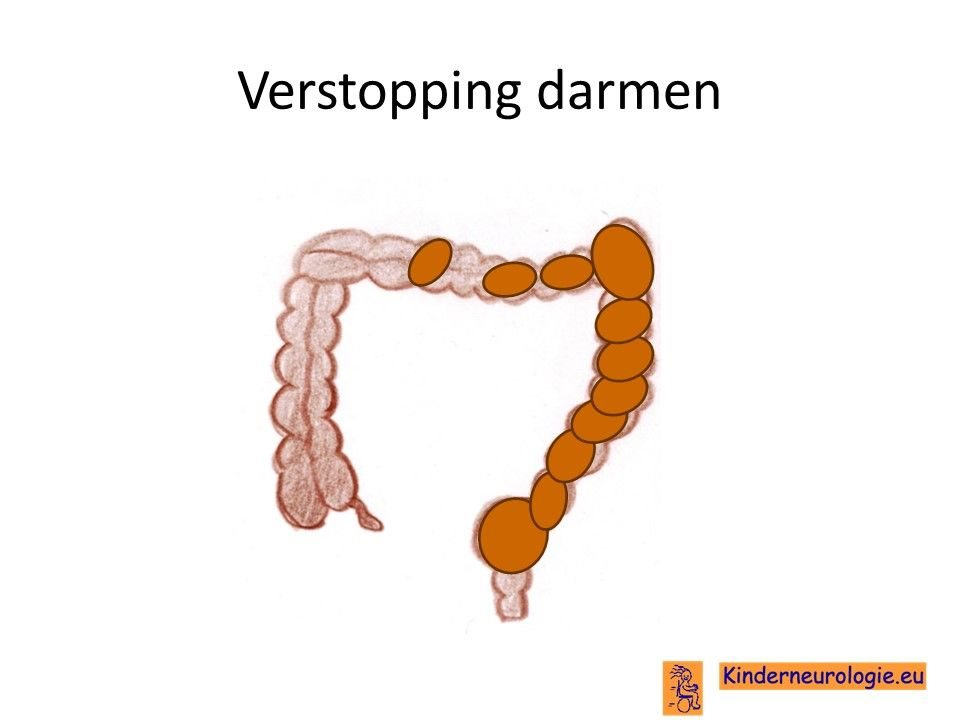
Verstopping van de darmen
Verstopping van de darmen komt vaak voor bij kinderen met dit syndroom. De ontlasting komt dan niet elke dag en is vaak hard waardoor kinderen moeite hebben met poepen. Dit kan buikpijnklachten geven en zorgen voor een bolle buik. Ook kan de eetlust hierdoor minder worden.
Niet ingedaald balletjes
Bij een groot deel van de jongentjes zitten de balletjes niet in het balzakje. Dit wordt cryptorchisme genoemd.

Huidafwijkingen
Kinderen met Smith-Kingsmore syndroom hebben vaak een wat gevoeligere droge huid. Kinderen zijn kwetsbaarder om last te krijgen van eczeem.
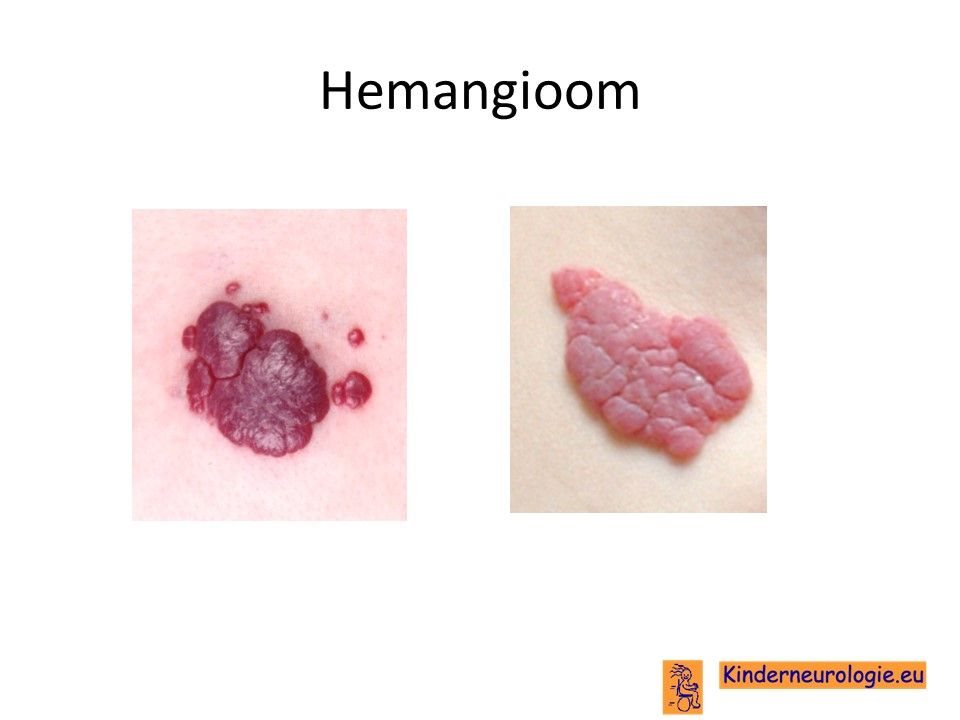
Een deel van de kinderen heeft ook enkele lichtbruine vlekken op de huid. Ook zichtbare uitgezette bloedvaatjes in de huid (hemangioom) kunnen voorkomen.
Blauwe plekken
Een deel van de kinderen krijgt gemakkelijk blauwe plekken na licht stoten, omdat deze kinderen een tekort een bloedplaatjes hebben. Ook kunnen frequente bloedneuzen hierop wijzen.
Vermoeidheid
Kinderen met het Smith-Kingsmore syndroom zijn vaak sneller vermoeid dan andere kinderen. Dit komt dat zij vaak meer tijd nodig hebben om alle informatie om hen heen te verwerken. Daarnaast zorgt de lagere spierspanning er voor dat kinderen meer energie moeten steken in het stabiel houden van hun gewrichten. Dit kost allemaal energie en energie kan maar een keer worden uitgegeven.
Slapen
Kinderen met het Smith-Kingsmore syndroom hebben vaak een grote slaapbehoefte. Zij zijn vaak van baby af aan al goede slapers, die goed kunnen inslapen en doorslapen. Zij hebben meer slaap nodig dan hun leeftijdsgenoten. Ook slaaptekort kan bijdragen aan vermoeidheid overdag. Een deel van de kinderen heeft moeite met inslapen. Slaaproblemen kunnen het gevolg zijn van nachtelijke epilepsie, het is goed hier alert op te zijn.
Scoliose
Een klein deel van de kinderen krijgt een milde verkromming van de wervelkolom. Een dergelijke verkromming wordt scoliose genoemd. Meestal hebben kinderen met Smith-Kingsmore syndroom hier geen last van.
Vatbaarheid voor infecties
Jonge kinderen met Smith-Kingsmore syndroom zijn vatbaarder voor het krijgen van luchtweg- en oorinfecties op jonge leeftijd.
Hoe wordt de diagnose Smith-Kingsmore syndroom gesteld?
Verhaal en onderzoek
Aan de hand van het verhaal van een kind met een groot hoofd en een lagere spierspanning kan worden vermoed dat er sprake is van een syndroom. Er bestaan meerdere syndromen die soortgelijke symptomen kunnen geven.
Aanvullend onderzoek zal nodig zijn om de juiste diagnose te stellen.
DNA-onderzoek
Door middel van bloedonderzoek kan gekeken worden of er een fout in het erfelijk materiaal gevonden kan worden. Dit kan gericht gebeuren wanneer er gedacht wordt aan het Smith-Kingsmore syndroom omdat dit in de familie voorkomt.
Tegenwoordig bestaat ook een genetische techniek, whole exome sequencing (WES) genoemd, waarbij in een keer een heleboel fouten in het DNA kunnen worden opgespoord. Zo kan de diagnose Smith-Kingsmore syndroom worden gesteld zonder dat er gericht aan gedacht wordt.
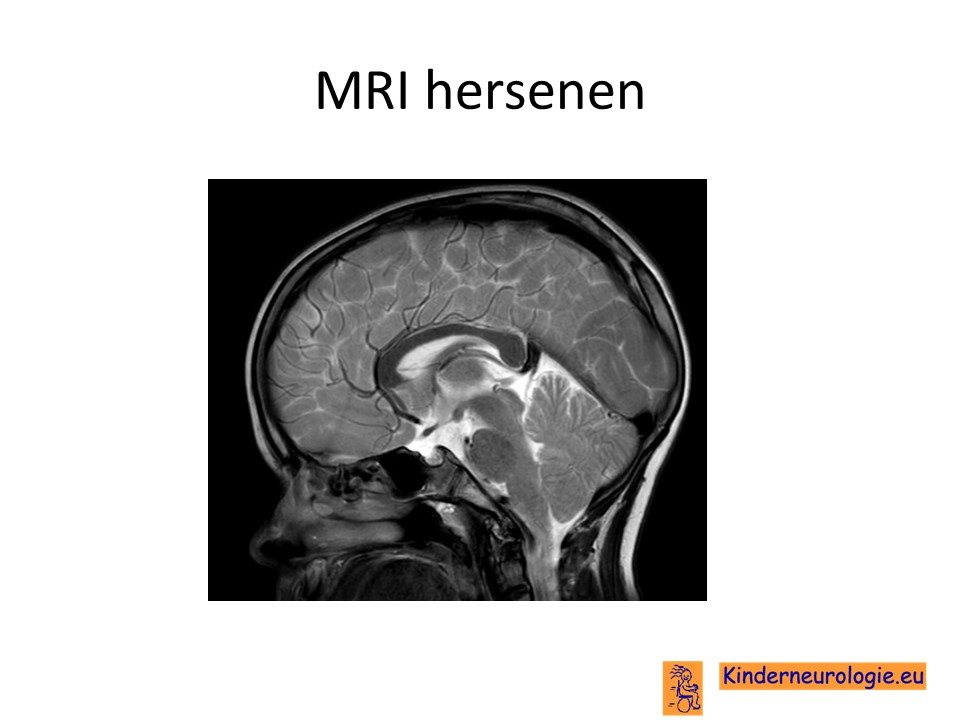
MRI van de hersenen
Kinderen met een groot hoofd zullen een MRI scan van de hersenen krijgen om te kijken wat de oorzaak is van het grote hoofd. Bij kinderen met Smith-Kingsmore syndroom is vaker te zien dat de hersenholtes iets groter zijn dan gebruikelijk, zonder dat er aanwijzingen zijn voor een waterhoofd. Ook kan er extra vocht aan de buitenkant van de hersenen zichtbaar zijn, dit wordt een benigne externe hydrocefalus genoemd. De hersenbalk kan dunner zijn dan gebruikelijk. Ook kan een deel van de hersenbalk ontbreken. De hersenen kunnen op een bepaalde plek anders zijn aangelegd dan gebruikelijk, dit wordt een corticale dysplasie genoemd. Soms komen meer windingen in de hersenschors voor, dit wordt een polymicrogyrie genoemd. Bij een klein deel van de kinderen zijn zogenaamde witte vlekken in de witte stof te zien. Ter plaatse is het geleidingslaagje rondom de zenuwen waarschijnlijk niet goed aangelegd. Ook kan het geleidingslaagje trager worden aangelegd dan gebruikelijk. De hersenstam kan dunner zijn dan gebruikelijk.
EEG
Kinderen met verdenking op epilepsieaanvallen krijgen vaak een EEG om te kijken van welk soort epilepsie er sprake is. Op het EEG worden vaak epileptiforme afwijkingen gezien. Deze afwijkingen zijn niet kenmerkend voor het Smith-Kingsmore syndroom, maar kunnen bij veel andere syndromen met epilepsie ook gezien worden.
Bloedonderzoek
Een deel van de kinderen heeft een verlaagd aantal bloedplaatjes in het bloed. Bloedonderzoek kan ook een verlaagde bloedsuiker waarde in het bloed aantonen. Soms is er sprake van een te lage waarde van de afweerstof IgA.
Oogarts
De oogarts kan beoordelen of er sprake is van een probleem met zien.

Kindercardioloog
De kindercardioloog kan beoordelen of er sprake is van een aangeboren hartafwijking.
Hoe wordt Smith-Kingsmore syndroom behandeld?
Geen genezing
Er bestaat geen behandeling die het Smith-Kingsmore syndroom kan genezen. De behandeling is er op gericht om de ontwikkeling van kinderen met Smith-Kingsmore syndroom zo goed mogelijk te laten verlopen en om eventueel complicaties van deze aandoening zo vroeg mogelijk op te sporen en te behandelen.
Afplakken oog
Bij kinderen die last hebben van scheelzien, is het vaak nodig om een oog af te plakken om te voorkomen dat een oog een zogenaamd lui oog wordt, waarmee kinderen niet meer goed kunnen zien. Een deel van de kinderen heeft een bril nodig om goed te kunnen zien.

KNO-arts
Bij kinderen met frequente middenoorinfecties kunnen buisjes nodig zijn om vocht achter het trommelvlies weg te laten lopen.
Voorkomen overgewicht
Kinderen met het Smith-Kingsmore syndroom krijgen tijdens de tienerleeftijd gemakkelijk last van overgewicht. Het is daarom belangrijk om al vanaf jonge leeftijd kinderen te leren niet te veel te snoepen. Een diëtiste kan adviezen geven voor goede voeding om overgewicht te voorkomen. En daarnaast te zorgen dat kinderen voldoende lichaamsbeweging krijgen.

Fysiotherapie
Een fysiotherapeut kan ouders tips en adviezen geven hoe ze hun kindje zo goed mogelijk kunnen stimuleren om er voor te zorgen dat de ontwikkeling zo optimaal als mogelijk verloopt.
Logopedie
Een logopediste kan tips en adviezen geven indien er problemen zijn met zuigen, drinken, kauwen of slikken. Sommige kinderen hebben baat bij een speciale speen (special need speen) waardoor het drinken uit de fles beter verloopt. Moeders kunnen borstvoeding kolven, zodat kinderen op deze manier toch borstvoeding als voeding kunnen krijgen via de fles.
Ook kan de logopediste helpen om de spraakontwikkeling zo goed mogelijk te stimuleren. Praten kan ook ondersteund worden door middel van gebaren of pictogrammen. Op die manier kunnen kinderen zich leren uitdrukken ook als ze nog geen woorden kunnen gebruiken.

Revalidatiearts
Een revalidatiearts coördineert de verschillende therapieën en adviseert ook over hulpmiddelen zoals bijvoorbeeld een aangepaste buggy, een rolstoel, steunzolen of aangepaste schoenen.
Ook is het mogelijk via een revalidatie centrum naar een aangepaste peutergroep te gaan en daar ook therapie te krijgen en later op dezelfde manier onderwijs te gaan volgen.
School
Het merendeel van de kinderen met Smith-Kingsmore syndroom volgt regulier onderwijs al dan niet met extra ondersteuning. Een deel van de kinderen volgt speciaal basisonderwijs van cluster 2 of 4. In het speciaal onderwijs zijn de klassen kleiner en kan meer begeleiding gegeven worden.
Orthopedagoog
Een orthopedagoog kan ouders tips en adviezen geven hoe om gaan met problemen met bijvoorbeeld boos worden of contact maken met andere kinderen.
Kinder- en jeugdpsychiater
Een kinder- en jeugdpsychiater kan advies geven hoe om te gaan met gedragsproblemen zoals ADHD of autisme. Soms is het nodig om gedragsregulerende medicatie zoals methylfenidaat voor ADHD of risperidon of aripiprazol voor prikkelovergevoeligheid te geven.
Behandeling epilepsie
Met behulp van medicijnen wordt geprobeerd om de epilepsieaanvallen zo veel mogelijk te voorkomen en het liefst er voor te zorgen dat er helemaal geen epilepsieaanvallen meer voorkomen. Soms lukt dit vrij gemakkelijk met een medicijn, maar bij een deel van de kinderen is het niet zo eenvoudig en zijn combinaties van medicijnen nodig om de epilepsie aanvallen zo veel mogelijk of helemaal niet meer te laten voorkomen.
Verschillende soorten medicijnen kunnen gebruikt worden om de epilepsie onder controle te krijgen, er bestaat geen voorkeursmedicijn voor de behandeling van epilepsie bij kinderen met Smith-Kingsmore syndroom.

Navelbreuk
Een navelbreuk heeft meestal geen behandeling nodig. Met het ouder worden, wordt de navelbreuk meestal spontaan kleiner.
Verstopping van de darmen
Het medicijn macrogol kan er voor zorgen dat de ontlasting soepel en zacht blijft en stimuleert de darmwand om actief te blijven. Hierdoor kunnen kinderen gemakkelijker hun ontlasting kwijt. Verder blijft het belangrijk om te zorgen dat kinderen voldoende vocht en vezels binnen krijgen en zo veel als kan bewegen. Soms zijn zetpillen nodig om de ontlasting op gang te krijgen.

Begeleiding
Een maatschappelijk werkende of psycholoog kan begeleiding geven hoe het hebben van deze aandoening een plaatsje kan krijgen in het dagelijks leven. Het kost vaak tijd voor ouders om te verwerken dat hun kind een syndroom heeft en om te gaan met de onzekerheden die horen bij het hebben van een syndroom.

Door middel van een oproepje op het forum van deze site kunt u proberen in contact te komen met andere kinderen en hun ouders/verzorgers die ook te maken hebben met het Smith-Kingsmore syndroom.
Wat is de prognose van kinderen met Smith-Kingsmore syndroom?
Zelfstandig leven
Het merendeel van de volwassenen met Smith-Kingsmore syndroom kan zelfstandig zijn of haar leven leiden zonder de hulp van anderen. Een deel van de volwassenen, met autistiforme kenmerken, heeft op volwassen leeftijd wel de hulp van een andere volwassene nodig tijdens het dagelijks leven.
Weinig bekend
Er is nog maar weinig bekend over volwassenen met het Smith-Kingsmore syndroom.
Levensverwachting
Er zijn geen gegevens bekend over de levensverwachting van kinderen en volwassenen met het Smith-Kingsmore syndroom. Er zijn tot nu toe geen aanwijzingen dat de levensverwachting van kinderen en volwassenen anders zou zijn die van kinderen en volwassenen zonder dit syndroom.
Kinderen
Volwassen met Smith-Kingsmore syndroom kunnen kinderen krijgen. Deze kinderen hebben 50% kans om zelf ook het Smith-Kingsmore syndroom te krijgen. Of deze kinderen evenveel, minder of meer symptomen zullen krijgen dan hun ouder valt van te voren niet te voorspellen.
Hebben broertjes of zusjes ook kans Smith-Kingsmore syndroom te krijgen?
Erfelijke ziekte
Het Smith-Kingsmore syndroom wordt veroorzaakt door een fout in het erfelijk materiaal. Wanneer een van de ouders zelf het Smith-Kingsmore syndroom heeft, dan hebben broertjes en zusjes 50% kan om zelf ook het Smith-Kingsmore syndroom te krijgen.
Wanneer de fout bij het kind zelf is ontstaan, dan is de kans erg klein dat een broertje of zusjes ook zelf het Smith-Kingsmore syndroom krijgt. Dit zou alleen kunnen wanneer de fout bij de vader in de zaadcellen of bij de moeder in de eicellen zit zonder dat zij dit in de andere lichaamscellen hebben. De kans hierop is 1-2%.
Een klinisch geneticus kan hier meer informatie over geven.
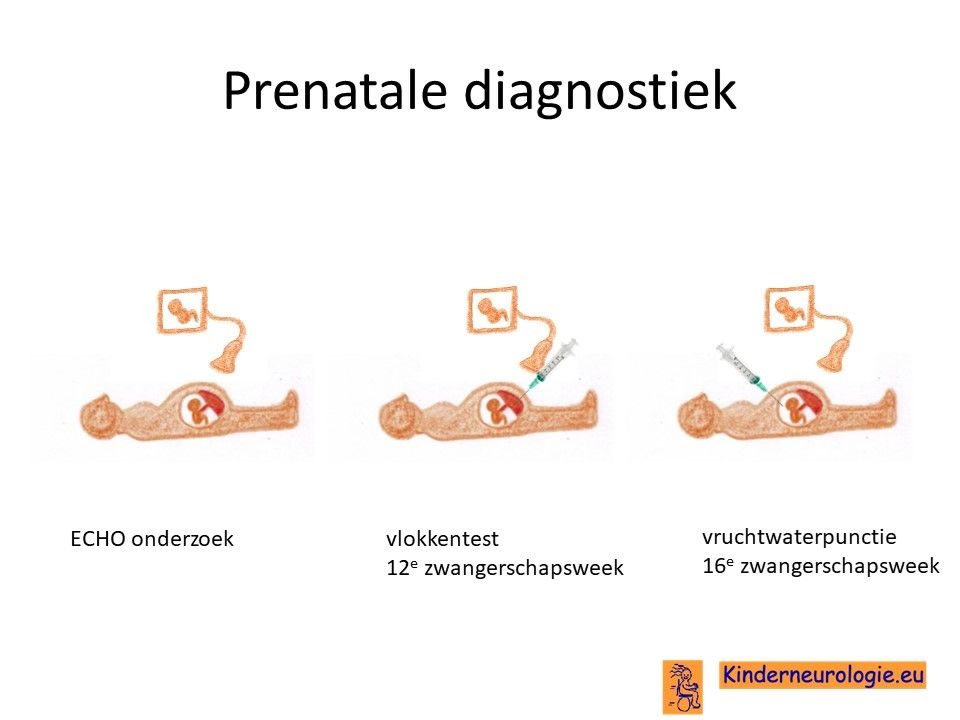
Prenatale diagnostiek
Door middel van een vlokkentest in de 12e zwangerschapsweek of een vruchtwaterpunctie in de 16e zwangerschapsweek bestaat de mogelijkheid om tijdens een zwangerschap na te gaan of een broertje of zusje ook deze aandoening heeft. Beide ingrepen hebben een klein risico op het ontstaan van een miskraam (0,5% bij de vlokkentest en 0,3% bij de vruchtwaterpunctie). De uitslag van deze onderzoeken duurt twee weken. Voor prenatale diagnostiek kan een zwangere de 8ste week verwezen worden door de huisarts of verloskundige naar een afdeling klinische genetica. Meer informatie over prenatale diagnostiek kunt u vinden op de website: www.pns.nl
Preïmplantatie Genetische Test (PGT)
Stellen die eerder een kindje hebben gehad met het Smith-Kingsmore syndroom, kunnen naast prenatale diagnostiek ook in aanmerking voor preïmplantatie genetische diagnostiek(PGD.) Bij PGD wordt een vrouw zwanger door middel van IVF (In Vitro Fertilisatie). De bevruchting vindt dan buiten het lichaam plaats, waardoor het zo ontstane pre-embryo onderzocht kan worden op het hebben van het Smith-Kingsmore syndroom. Alleen embryo’s zonder de aanleg voor het Smith-Kingsmore syndroom, komen in aanmerking voor terugplaatsing in de baarmoeder. Voor meer informatie zie www.pgdnederland.nl.
Wilt u ook uw verhaal kwijt, dat kan: verhalen kunnen gemaild worden via via contact en zullen daarna zo spoedig mogelijk op de site worden geplaatst. Voor meer informatie zie hier.
Heeft u foto's die bepaalde kenmerken van deze aandoening duidelijk maken en die hier op de website mogen worden geplaatst, dan vernemen wij dit graag via contact.
Links
https://smithkingsmore.org
(Engelstalige website over het Smith Kingsmore syndroom)
Referenties
- mTOR mutations in Smith-Kingsmore syndrome: Four additional patients and a review. Gordo G, Tenorio J, Arias P, Santos-Simarro F, García-Miñaur S, Moreno JC, Nevado J, Vallespin E, Rodriguez-Laguna L, de Mena R, Dapia I, Palomares-Bralo M, Del Pozo Á, Ibañez K, Silla JC, Barroso E, Ruiz-Pérez VL, Martinez-Glez V, Lapunzina P. Clin Genet. 2018;93:762-775.
- Smith-Kingsmore syndrome: A third family with the MTOR mutation c.5395G>A p.(Glu1799Lys) and evidence for paternal gonadal mosaicism. Moosa S, Böhrer-Rabel H, Altmüller J, Beleggia F, Nürnberg P, Li Y, Yigit G, Wollnik B. Am J Med Genet A. 2017;173:264-267
Laatst bijgewerkt: 6 augustus 2022 voorheen: 15 augustus 2021, 25 september 2019 en 23 februari 2019
Auteur: JH Schieving