
Wat is een non-ketotische hyperglycinemie?
Een non-ketotische hyperglycinemie is een stofwisselingsziekte waardoor er te veel van het stofje glycine in het lichaam aanwezig is, waardoor de hersenen niet goed functioneren.
Hoe wordt een non-ketotische hyperglycinemie ook wel genoemd?
Bij deze ziekte is er een te veel aan het stofje glycine. Hier verwijst de term hyper in hyperglycinemie naar. Te veel aan glycine is schadelijk voor de hersenen. De hersenen functioneren daarom minder goed.
Er is bij deze aandoening geen sprake van ketose in het bloed, vandaar de woorden non-ketotisch. Non-ketotische hyperglycinemie wordt wel afgekort met de letters NKH.
Glycine-encefalopathie
Een non-ketotische hyperglycinemie wordt ook wel een glycine-encefalopathie genoemd.Het niet goed functioneren van de hersenen wordt een encefalopathie genoemd.
Verschillende types
Er bestaan verschillende types NKH. De indeling is gemaakt naar het moment waarop kinderen hun eerste klachten krijgen. De meeste kinderen krijgen kort na de geboorte hun eerste klachten. Dit wordt het klassieke type genoemd.
Er zijn ook kinderen waarbij de eerste klachten in de eerste vier levensweken ontstaan, dit wordt het neonatale type genoemd. Wanneer de eerste klachten in het eerste levensjaar ontstaan wordt gesproken van het infantiele type. Wanneer de eerste klachten pas na de leeftijd van een jaar ontstaan wordt gesproken van een laat begin van de ziekte.
Meestal is het zo dat het ziektebeeld ernstiger verloopt naarmate de klachten op jongere leeftijd ontstaan.

Hoe vaak komt een non-ketotische hyperglycinemie voor bij kinderen?
Een non-ketotische hyperglycinemie is een zeldzame ziekte. Geschat wordt dat het bij één op de 50.000 kinderen voorkomt.

Bij wie komt een non-ketotische hyperglycinemie voor?
Een non-ketotische hyperglycinemie geeft meestal al kort na de geboorte ernstige klachten. Soms ontstaan de eerste symptomen pas op peuter/kleuter leeftijd. Kinderen met voorouders uit Finland hebben een grotere kans om NKH te krijgen, omdat in Finland meer mensen drager zijn van een fout in het DNA die NKH kan geven.
Zowel jongens als meisjes kunnen een non-ketotische hyperglycinemie krijgen. Om nog onbekende redenen hebben jongens meestal minder klachten dan meisjes.
Wat is de oorzaak van een non-ketotische hyperglycinemie?
Fout in het erfelijk materiaal
Non-ketotische hyperglycinemie wordt veroorzaakt door een foutje in het erfelijk materiaal.
Er zijn drie verschillende fouten bekend die allemaal NKH kunnen veroorzaken. De plaats van de fout op het erfelijk materiaal worden GLDC-gen, AMT-gen en GCSH-gen genoemd. De meeste kinderen hebben een fout in het GLDC-gen.
Het GLDC-gen ligt op chromosoom 9, het AMT-gen op chromosoom 3 en het GCSH-gen op chromosoom 16.

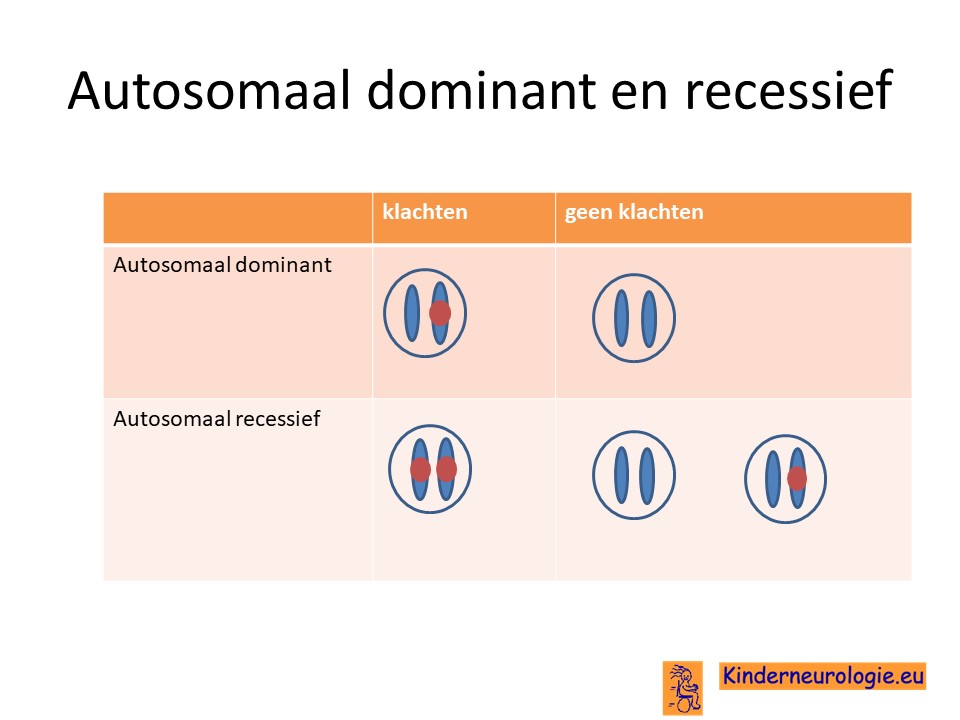
Autosomaal recessieve ziekte
Non-ketotische hyperglycinemie is een erfelijke ziekte. Dat wil zeggen dat kinderen pas klachten wanneer twee chromosomen van het zelfde nummer op dezelfde plaats een fout bevatten.
Dit in tegenstelling tot een autosomaal dominante aandoening. Hierbij krijgen kinderen al last van een ziekte wanneer een van de twee chromosomen een afwijking bevat.
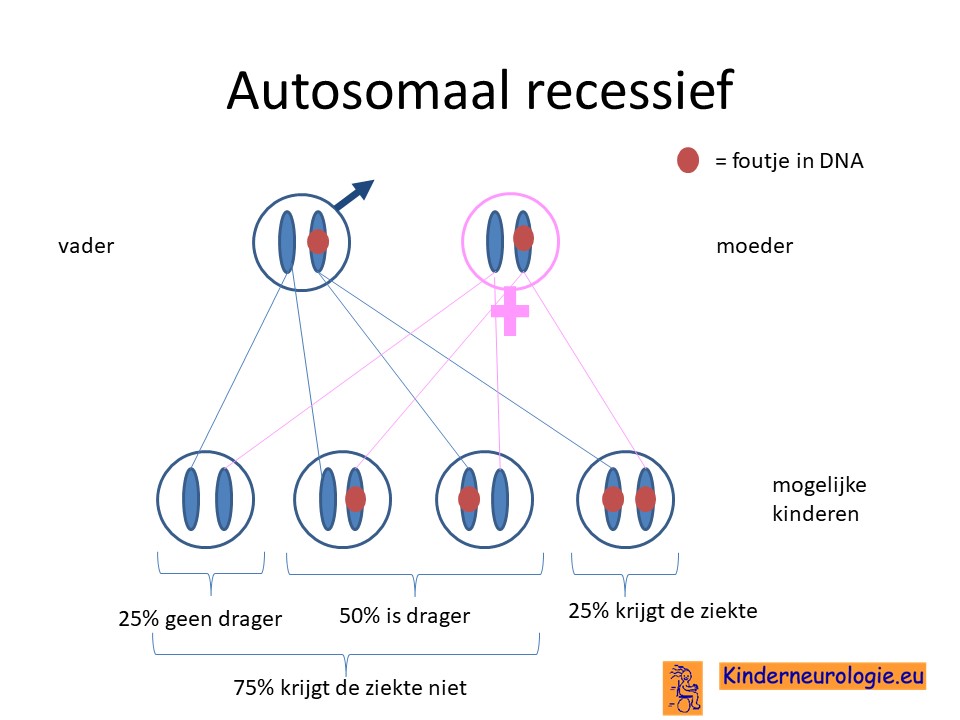
Ouders drager
Bij een autosomaal recessieve ziekte zijn beide ouders vaak drager van een afwijkend gen op een chromosoom. Omdat de ouder zelf nog een ander chromosoom zonder fout, heeft de ouder zelf geen klachten. De meeste ouders weten dan ook niet dat ze drager zijn.
Wanneer twee ouders drager zijn, dan hebben zij samen 25% kans om een kindje te krijgen met NKH. Wanneer ouders in de verte familie van elkaar zijn, is de kans groter dat zij drager zijn van een zelfde soort fout in het DNA.
Afwijkend eiwit
Als gevolg van het foutje in het erfelijke materiaal wordt een bepaald eiwit niet aangemaakt bij kinderen met non-ketotische hyperglycinemie Dit eiwit heeft glycine-decarboxylase of ook wel glycine klievings (cleavage) eiwit genoemd. Dit eiwit speelt een belangrijke rol bij de afbraak van glycine in de energiefabriekjes van het lichaam (mitochondriëen). Dit eiwit bestaat uit vier onderdelen die aangeduid worden met de letters P, T, H en L. Drie op de vier kinderen met NKH heeft een afwijkend P-eiwit. Een deel van de kinderen heeft een afwijkend T-eiwit. Een afwijkend H-eiwit is uiterst zeldzaam.
Het GLDC-gen zorgt voor de aanmaak van het P-eiwit. Het AMT-gen voor de aanmaak van het T-eiwit en het GCSH-gen voor de aanmaak van het H-eiwit.
Afwijkende aanleg van de hersenen
Omdat het stofje glycine niet goed afgebroken en opgeruimd kan worden, is er te veel glycine aanwezig in de hersenen. Hierdoor kunnen de hersenen zich niet normaal ontwikkelen. De ontwikkeling van de hersenen verloopt tijdens de zwangerschap al anders dan gebruikelijk.
De hersenen zijn vaak kleiner dan gebruikelijk. De verbinding tussen de rechter en de linker hersenhelft (het corpus callosum ) is vaak niet aangelegd. Ook krijgen de hersencellen niet een beschermend geleidingslaagje, het myelinelaagje. Hierdoor werken de hersencellen traag en raken ze gemakkelijk beschadigd.

Glycine
Glycine wordt gebruikt als boodschapperstofje in de hersenen. Dit boodschapper stofje wordt op verschillende plaatsen in de hersenen gebruikt en heeft verschillende functies. In de hersenstam en in het ruggenmerg heeft glycine een dempende werking op de zenuwcellen. Dit verklaart waarom kinderen met een hyperlgycinemie vaak sloom en slaperig zijn en gemakkelijk last van de hik krijgen. In de grote hersenen wis glycine een co-activator van de NMDA-receptor en heeft het juist een activerend effect. Dit verklaard waarom kinderen met een hyperglycinemie last van epilepsie krijgen. Ook kan overmatig activeren van de NMDA receptor zorgen voor schade aan zenuwcellen, waardoor zenuwcellen verloren kunnen gaan.

Beschadiging van de hersenen
Het te veel aan glycine in de hersenen zorgt er ook voor dat bepaalde zenuwcellen die het stofje glutamaat gebruiken als boodschapperstofje te hard gaan werken. Te veel aan het stofje glutamaat zorgt voor beschadiging van de hersencellen die wel normaal aangelegd zijn.

Afwijkende lever
Het glycine-decarboxylase eiwit speelt ook een belangrijke rol in de lever. Toch komen problemen met de lever niet veel voor, mogelijk vallen ze niet goed op, omdat de problemen met de hersenen veel ernstiger zijn.
Wat zijn de symptomen van een non-ketotische hyperglycinemie?
Klassieke type
Het klassieke type van NKH komt het meeste voor. Bij dit type zijn de symptomen al bij de geboorte aanwezig of ontstaan ze in de eerste uren na de geboorte.
Jouw kind is uniek
Bedenk dat onderstaande symptomen kunnen voorkomen bij jouw kind, maar ook niet allemaal zullen voorkomen. Jouw kind is uniek en veel meer dan een kind met deze aandoening. Het lezen van mogelijke symptomen die kunnen voorkomen, kan ouders het gevoel geven dat er alleen maar aandacht is voor de beperkingen van het kind. Dat is zeer zeker niet de bedoeling. Jouw kind is bijvoorbeeld lief, grappig, gevoelig, gezellig,sociaal, vindingrijk, nieuwsgierig, ondeugend, enthousiast,een zonnestraaltje, creatief en/of innemend en dat vind je niet terug in onderstaande symptomen die kunnen horen bij dit syndroom. Dat kan ook niet, want die eigenschappen maken jouw kind nu eenmaal uniek. Blijf daar vooral naar kijken en zie deze symptomen meer als achtergrondinformatie die je kunnen helpen om te begrijpen wat er met je kind aan de hand zou kunnen zijn wanneer jouw kind zich anders ontwikkelt of ergens last van heeft. Deze informatie kan jullie als ouders en hulpverleners een handvat geven wat hiervoor een mogelijke verklaring kan zijn.

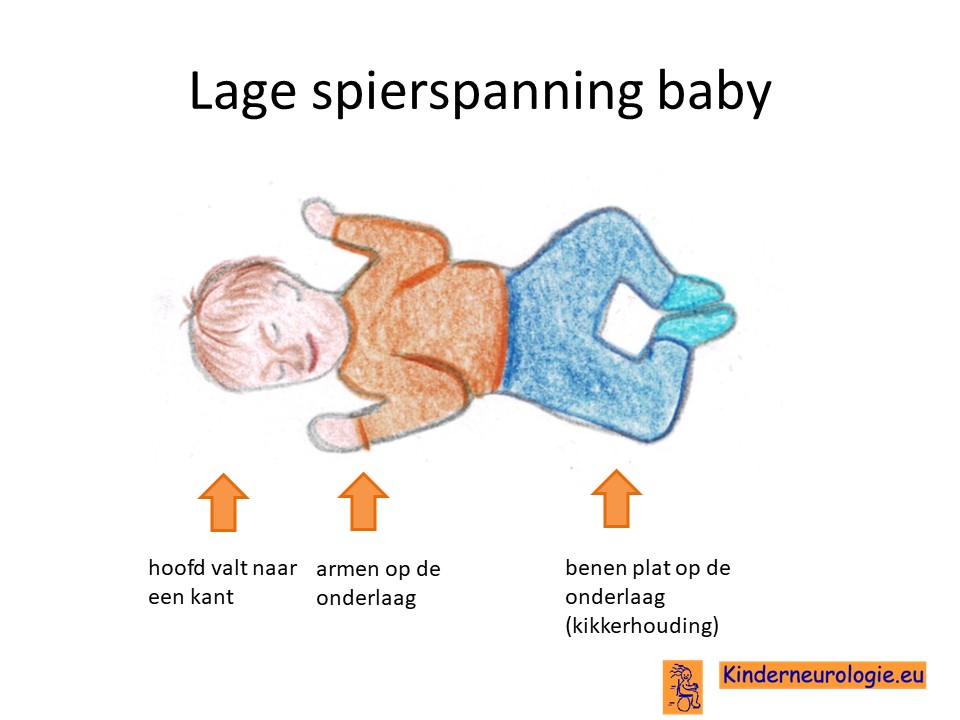
Lage spierspanning
Baby’s met NKH hebben vaak een lage spierspanning. Ze voelen vaak erg slap aan en moeten goed ondersteund worden wanneer ze gedragen worden. Baby’s kunnen een hoofd niet goed overeind houden.
Sloom
Baby’s met NKH zijn vaak erg sloom. Ze slapen meestal veel en zijn moeilijk wakker te krijgen. Wanneer ze wakker zijn, dan zijn ze niet actief. Hun armen en benen liggen stillen en bewegen nauwelijks. Wanneer kinderen hun ogen open hebben, dan hebben ze vaak een afwezige blik.
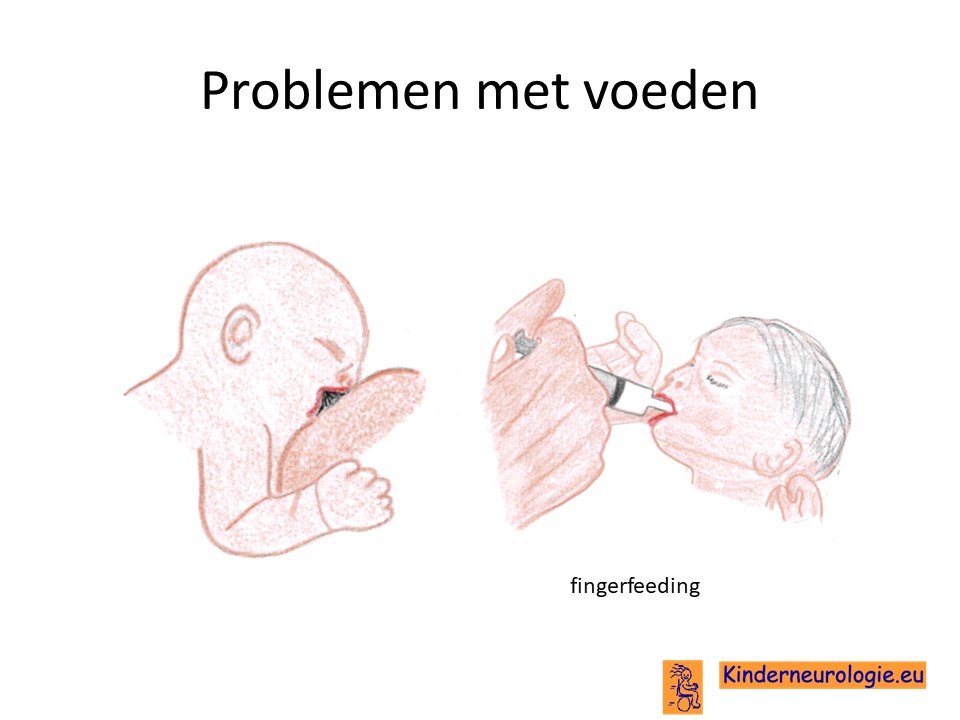
Problemen met drinken
Baby’s met een non-ketotische hyperglycinemie hebben vaak te weinig kracht en energie om te kunnen drinken uit de borst of uit de fles. Vaak zal het nodig zijn om kinderen voeding te geven via een sondeslangetje.
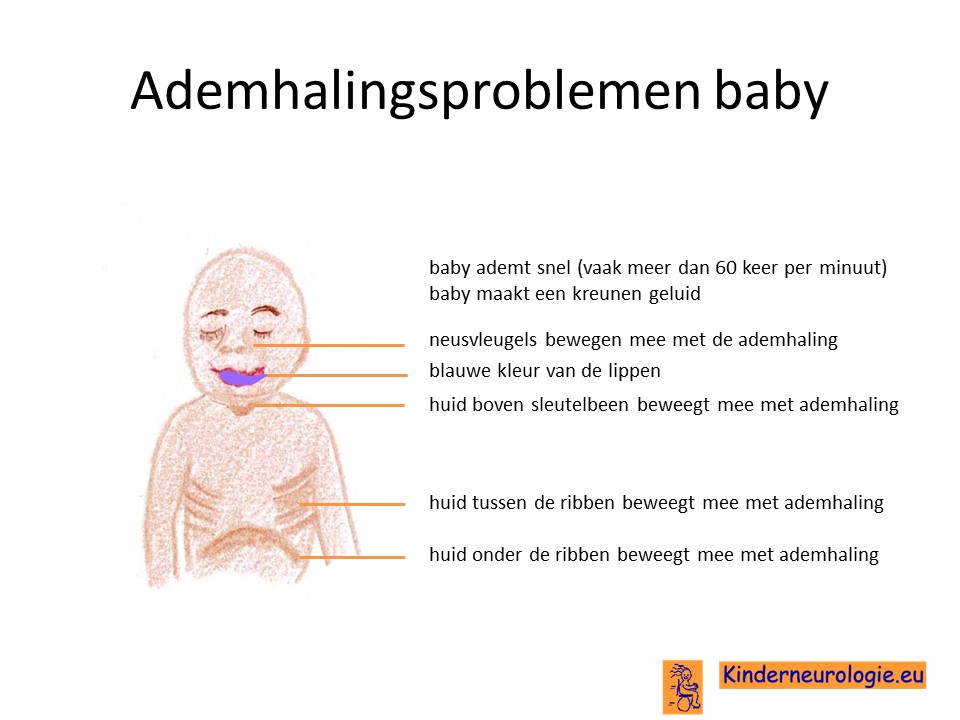
Ademhalingsproblemen
Kinderen met NKH ademen vaak erg oppervlakkig. Ook stoppen ze regelmatig met ademhalen. Dit worden apneu’s genoemd. Omdat kinderen niet op een goede manier ademhalen, hebben ze vaak beademing nodig om er voor te zorgen dat het lichaam voldoende zuurstof binnen krijgt.
Hik
Een deel van de kinderen met NKH heeft last van hardnekkige hik. Moeders herkennen dit vaak van tijdens de zwangerschap.
Zacht huilen
Kinderen met NKH huilen vaak zacht en klagelijk.
Schokkende bewegingen
Wanneer kinderen met NKH worden aangeraakt, dan lijken ze hierop te reageren met een heftige schrikbeweging. Beide armen en benen bewegen heftig op een aanraking. Deze beweging wordt een myoclonie genoemd.
Ook andere vormen van extra bewegingen, zoals dansende bewegingen (chorea), trillende beweging (tremor) of een afwijkende stand van een bijvoorbeeld een arm (dystonie) kunnen voorkomen bij kinderen met een NKH. Sommige kinderen overstrekken zichzelf.

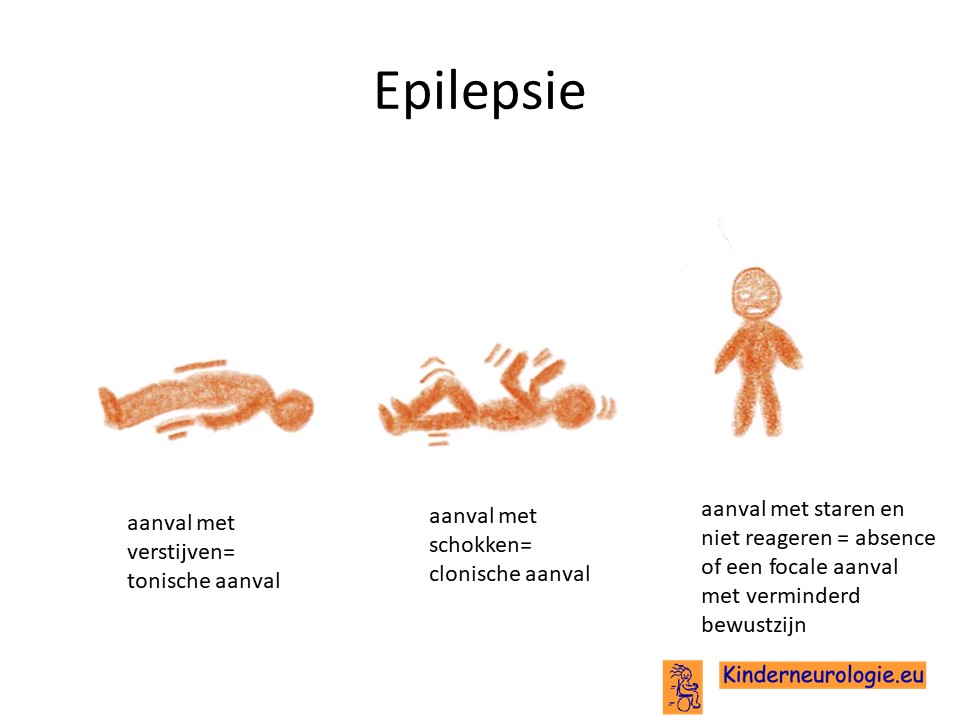
Epilepsie
Veel kinderen met NKH krijgen epilepsie-aanvallen. Deze aanvallen kunnen heel onopvallend zijn wanneer kinderen baby zijn. Op latere leeftijd komen andere type epilepsie aanvallen voor, die meer opvallen zoals schokkende bewegingen (clonische aanvallen) of een algehele verstijving van armen en/of benen (tonische aanvallen). Het is vaak heel moeilijk om deze epilepsie-aanvallen met behulp van medicijnen onder controle te krijgen.
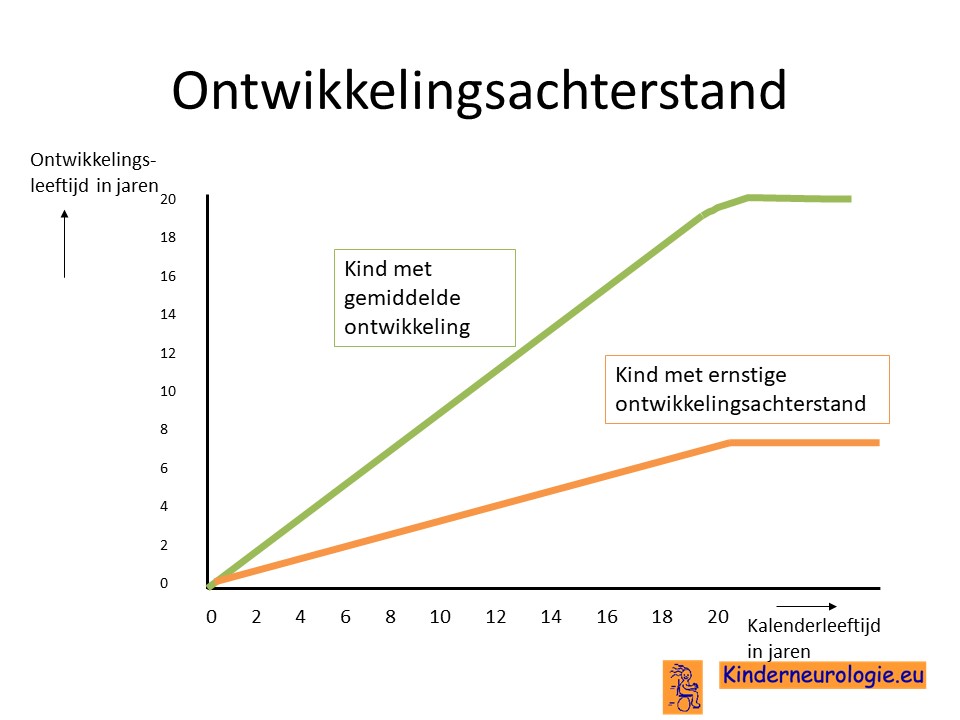
Ontwikkelingsachterstand
Het is voor kinderen met NKH heel moeilijk om stappen voorwaarts in de ontwikkeling te maken. Voor kinderen met epilepsie is dit nog moeilijker dan voor kinderen zonder epilepsie. Voor de kinderen die tot nu toe bekend zijn met het syndroom is het te moeilijk gebleken om zelfstandig te leren zitten, staan of lopen. Kinderen hebben een rolstoel nodig om zich te kunnen verplaatsen.
Ook het leren praten is moeilijk voor kinderen met de klassieke vorm van NKH.
Spasticiteit
Bij het ouder worden krijgen kinderen met NKH geleidelijk aan last van spasticiteit in de armen en benen. De armen en benen worden dan steeds stijver.Hierdoor wordt het bewegen van de armen en benen lastiger. De benen kunnen de neiging krijgen voor elkaar langs te kruizen, waardoor verschonen moeilijk wordt. Aanvallen met plostelinge toename van spierspanning kunnen voorkomen, dit wordt een spasme genoemd. Spasmes komen vooral voor bij vermoeidheid, ziek zijn, schrik of stress.

Autistiforme kenmerken
Kinderen met NKH hebben vaker autistiforme kenmerken. Kinderen zijn meer in zich zelf gekeerd en hebben niet zo’n behoefte aan contact met andere mensen. Zij leven in een eigen wereld. Het maken van oogcontact vinden kinderen vaak moeilijk.
Kinderen met autistiforme kenmerken houden vaak van een vaste voorspelbare structuur in de dag. Zij vinden het lastig wanneer hiervan wordt afgeweken. Ook onverwachte gebeurtenissen zijn moeilijk. Kinderen kunnen door onverwachte gebeurtenissen heel boos of juist heel verdrietig worden, omdat ze niet goed weten hoe ze hier mee om moeten gaan.Kinderen met autisme vinden het vaak moeilijk om emoties van andere mensen te kunnen begrijpen en weten niet goed hier op te reageren. Samen spelen en samen plezier hebben is vaak moeilijk voor kinderen met autisme.
Ook hebben kinderen vaak voorkeur voor bepaald speelgoed of een bepaalde hobby waar ze zich heel lang mee kunnen vermaken.

Klein hoofd
Het hoofd van kinderen met een NKH groeit minder goed. Daardoor krijgen kinderen verhoudingsgewijs een kleiner hoofd.

Waterhoofd
Een klein deel van de kinderen krijgt juist last van een waterhoofd. Hun hoofd groeit te snel en daarnaast kunnen klachten ontstaan van braken en ogen die naar beneden gericht staan. Dit worden zogenaamde sunset eyes genoemd.

Problemen met zien
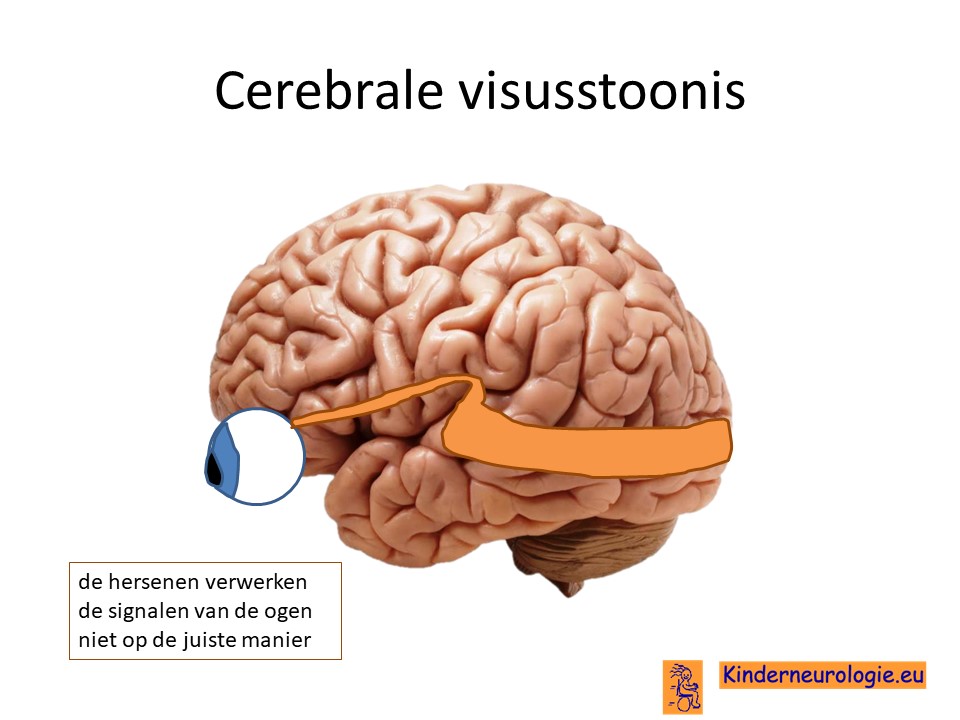
Een deel van de kinderen met dit syndroom is slechtziend doordat de hersenen de signalen van de ogen niet goed verwerken.
Kinderen zijn daardoor slechtziend terwijl er met hun ogen niets aan de hand is.Vaak maken kinderen weinig oogcontact. Dit wordt een cerebrale visusstoornis genoemd, het probleem zit in de hersenen (cerebrum). Het wordt ook wel afgekort als CVI ( naar de Engelse termen cerebral visual impairment, impairment is beperking)
Kwijlen
Kinderen met hNKH hebben gemakkelijker last van kwijlen. Dit komt door slapheid van de spieren in het gezicht en rondom de mond, waardoor het speeksel gemakkelijk uit de mond loopt. Dit is namelijk de gemakkelijkste weg voor het speeksel, de andere optie van het doorslikken van speeksel kost bewuste aandacht van het kind totdat doorslikken van speeksel geautomatiseerd is. Dit is voor kinderen met dit syndroom moeilijker om aan te leren.
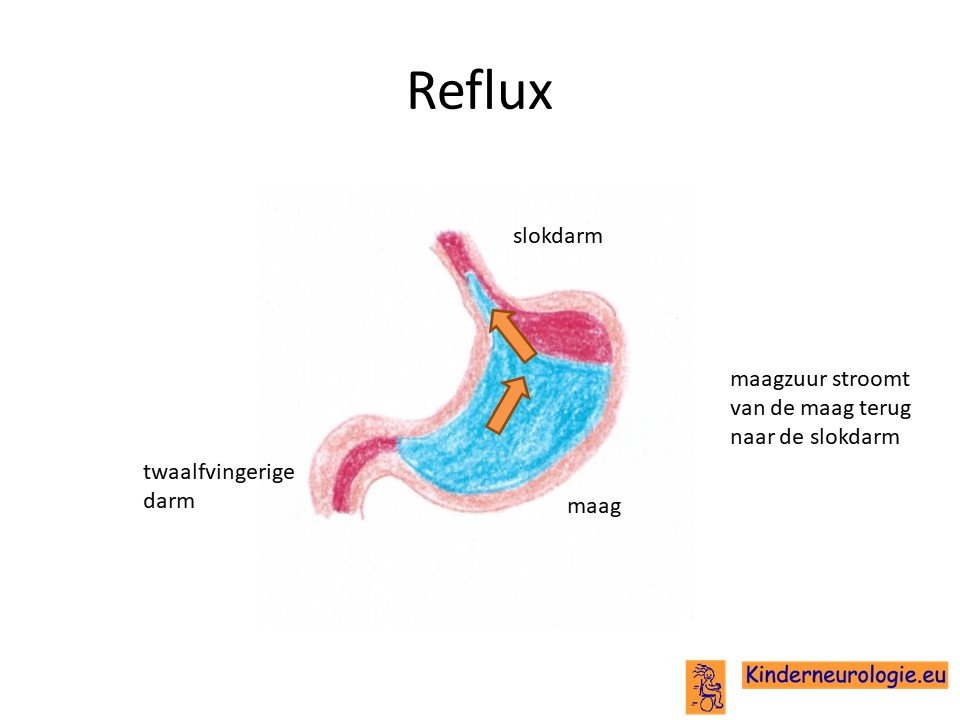
Reflux
Kinderen met heNKH hebben heel vaak last van het terugstromen van voeding vanuit de maag naar de slokdarm. Dit wordt reflux genoemd. Omdat de maaginhoud zuur is, komt het zuur zo ook in de slokdarm, soms zelfs ook in de mond. Dit zuur kan zorgen voor pijnklachten, waardoor kinderen moeten huilen en soms ook niet willen eten. Ook kan het maken dat kinderen moeten spugen.
Door het zuur kan de slokdarm geïrriteerd en ontstoken raken. Wanneer dit niet tijdige ontdekt en behandeld wordt, kan dit zorgen voor het spuug met daarin bloedsliertjes.
Verhoogde bloeddruk in de longen
Een deel van de kinderen krijgt een verhoogde bloeddruk in de longen. Dit kan klachten van kortademigheid geven.

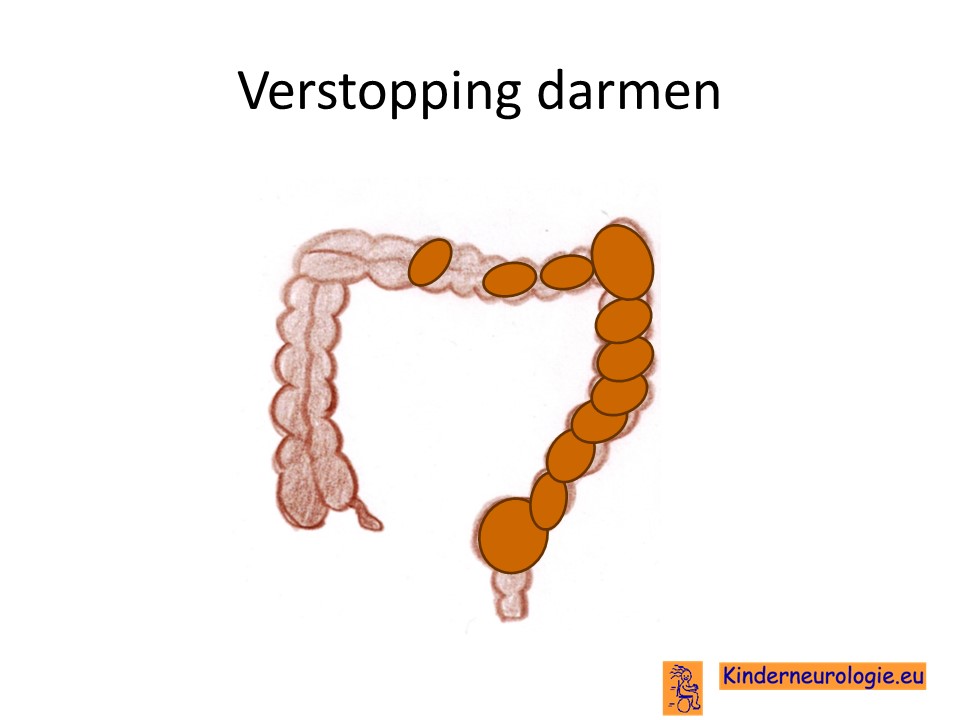
Verstopping van de darmen
Verstopping van de darmen komt vaak voor bij kinderen met NKH. De ontlasting komt dan niet elke dag en is vaak hard waardoor kinderen moeite hebben met poepen. Dit kan buikpijnklachten geven en zorgen voor een bolle buik. Ook kan de eetlust hierdoor minder worden.
Zindelijkheid
Voor een klein deel van de kinderen met NKH is het haalbaar om zindelijk te worden, voor een groot deel van de kinderen is dit niet haalbaar.

Scoliose
Een deel van de kinderen met dit syndroom krijgt een zijwaartse verkromming van de rug. Dit wordt een scoliose genoemd. Van een milde scoliose zullen kinderen zelf geen last hebben. Toename van de scoliose kan zorgen voor het ontstaan van pijnklachten in de rug en problemen met zitten en staan.
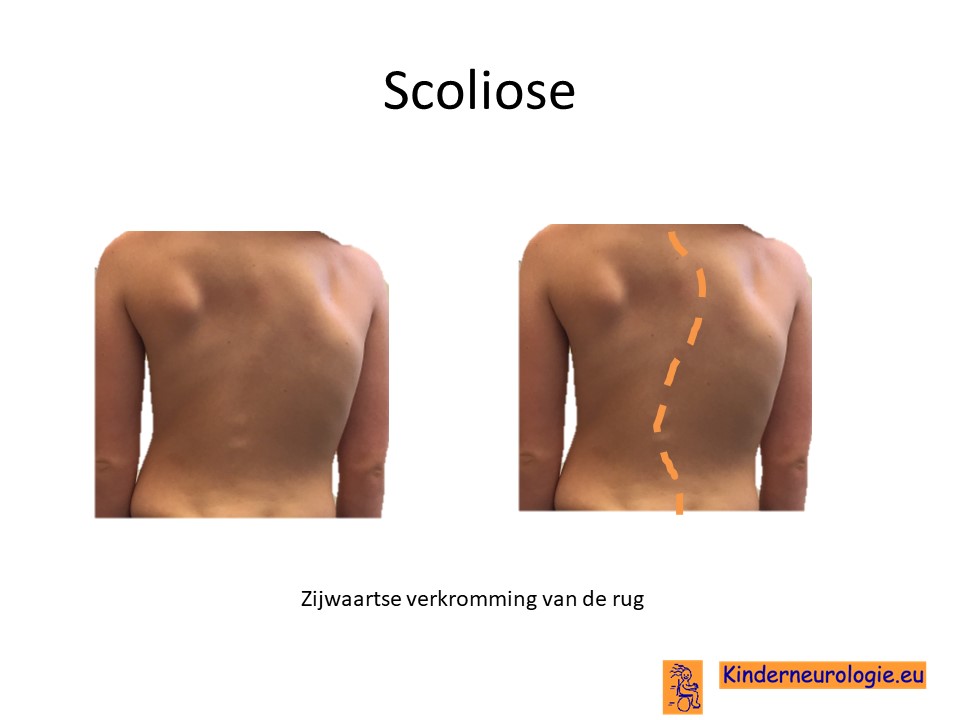
Heupdysplasie
Heupdysplasie komt vaker voor bij kinderen met dit syndroom. Hierbij is de heupkom niet goed ontwikkeld, waardoor de heupkop gemakkelijker uit de heupkom schiet.

Infantiele type
Een klein deel (een op de vijf kinderen met NKH) van de kinderen met een non-ketotische hyperglycinemie ontwikkelt zich tijdens de eerste levensmaanden normaal. Vanaf de leeftijd van gemiddeld zes maanden staat de ontwikkeling stil en krijgen kinderen epileptische aanvallen. De ontwikkeling gaat daarna meestal langzaam verder. De meeste kinderen met dit type zullen niet zelfstandig leren lopen. Zij hebben last van stijfheid (spasticiteit) van de armen en benen en ook last van bewegingsonrust (chorea) Ook krijgen zij last van de bovengenoemde schokkende bewegingen en epilepsie-aanvallen die moeilijk behandelbaar zijn. Vaak zijn er ook gedragsproblemen in de zin van druk gedrag en verminderde concentratie. Een deels van deze kinderen is heel onrustig in de manier van bewegen. Problemen met het evenwicht komen veel voor.
Atypische vormen
Bij nog een kleiner deel van de kinderen ontstaan de eerste klachten pas op latere leeftijd. Deze kinderen hebben meestal minder klachten. Vaak zijn er leerproblemen, gedragsproblemen en hebben deze kinderen ook epilepsie. Opvallend is dat kinderen tijdens koorts vaak niet goed omhoog en omlaag kunnen kijken, dit wordt een blikparese genoemd. Daarnaast hebben ze tijdens de koorts vaak last van bewegingsonrust, dit wordt chorea genoemd.
Kinderen met deze vorm krijgen vaak problemen met zien omdat hun oogzenuw steeds slechter gaat functioneren.
Hoe wordt de diagnose non-ketotische hyperglycinemie gesteld?
Verhaal en onderzoek
Aan de hand van het verhaal van een baby die na de geboorte erg slap en sloom is met een oppervlakkige ademhaling en ademstilstanden kan vermoed worden dat er iets met de hersenen van de baby aan de hand is. Er is nader onderzoek nodig om vast te stellen dat er sprake is van NKH. Ook andere stofwisselingsziektes en andere ziektes van de hersenen kunnen soort gelijke klachten geven.
Stofwisselingsonderzoek
Met behulp van bloed en urine kan gekeken worden of er problemen zijn met de stofwisseling. Bij kinderen met een non-ketotische hyperglycinemie is het stofje glycine in het bloed vaak verhoogd. Soms echter niet, dan zou de diagnose gemist kunnen worden.

Ruggenprik
Onderzoek van liquor (het vocht rondom de hersenen en het ruggenmerg) toont meestal wel een verhoogd glycine gehalte aan. Om dit vocht te verkrijgen is een ruggenprik nodig.
De verhouding tussen de hoeveelheid glycine in de liquor en glycine in het bloed is bij kinderen met een NKH meer dan 0,08. Een verhouding groter dan 0,15 geeft een grotere kans op een ernstig beloop van de ziekte, net als een glycine waarde hoger dan 116,5 umol/liter in het hersenvocht.
Bij kinderen met andere stofwisselingsziekten zoals organisch zuur syndroom kan het glycine in het bloed ook verhoogd zijn, maar meestal is het glycine in het hersenvocht verhoudingsgewijs niet verhoogd.
Glycine in bloed en liquor kan ook verhoogd zijn bij andere ziektes van de hersenen zoals hersenschade door zuurstoftekort. Niet automatisch betekent een verhoogd glycine gehalte in het hersenvocht dus dat er sprake is van een NKH.
Het blijkt dat bij kinderen met NKH de waardes van serine vaak verlaagd zijn en die van threonine vaak verhoogd. De verhouding tussen glycine en serine kan enige informatie geven over de ernst van het ziektebeloop van de NKH, hoe hoger deze waarde, hoe ernstiger bij de meeste kinderen het ziektebeloop.

Leverbiopt
Wanneer met bovenstaand stofwisselingsonderzoek onvoldoende duidelijk is dat er sprake is van een non-ketotische hyperglycinemie dan kan onderzoek verricht worden op een stukje uit de lever. Op deze manier kan gekeken worden of het eiwit wat zorgt voor de afbraak van glycine aanwezig is in de lever. Bij kinderen met een non-ketotische hyperglycinemie ontbreekt dit eiwit meestal helemaal.
DNA-onderzoek
Door middel van bloed onderzoek kan het DNA onderzocht worden. Op deze manier kan gekeken worden of er een fout in het erfelijk materiaal te vinden is die past bij de diagnose non-ketotische hyperglycinemie. Het lukt niet om bij alle kinderen een fout aan te tonen in het erfelijk materiaal. Waarschijnlijk zijn nog niet alle foutjes bekend.
EEG
Wanneer bij baby’s met NKH een hersenfilmpje (EEG) wordt gemaakt dan is daarop een speciaal patroon te hersenen. Dit patroon wordt een burst-supressie patroon genoemd.
Te zien is dat er af en toe een elektrische ontlading in de hersenen aanwezig is, gevolgd door een periode waarin de hersenen nauwelijks activiteit laten zien. Dit is een heel ernstig afwijkend patroon wat wijst op een ernstig probleem van de hersenen. Het wordt gezien bij NKH maar kan ook bij andere ernstige hersenaandoeningen gezien worden.

MRI van de hersenen
Wanneer er bij kinderen met NKH een MRI scan van de hersenen wordt gemaakt, dan is vaak te zien dat de hersenen onderontwikkeld zijn. Vaak zijn de hersenen te klein en ontbreekt de hersenbalk (corpus callosum) of soms een deel van de kleine hersenen. Ook is te zien dat de hersenen nog erg onrijp zijn, omdat er geen geleidingslaagje (myeline) rondom de hersencellen aanwezig is.
Met een speciale MRI techniek, MR-spectroscopie kan bij een deel van de kinderen aangetoond worden dat er te veel glycine in de hersenen aanwezig is.
Wanneer vervolg MRI scans gemaakt worden, is vaak te zien dat de hersenen van kinderen geleidelijk aan steeds kleiner worden door verlies van hersencellen.

Oogarts
Kinderen met dit syndroom worden altijd een keer gezien door de oogarts om te kijken of er afwijkingen zichtbaar zijn aan de ogen.

ECHO hart
De kindercardioloog kan door middel van een ECHO van het hart beoordelen of er sprake is van een verhoogde bloeddruk in de longen (pulmonale hypertensie).
Foto van de rug
Door middel van een röntgenfoto van de rug kan beoordeeld worden of er sprake is van een scoliose. Door het meten van de zogenaamde Cobbse hoek wordt de ernst van de scoliose weer gegeven.

Hoe wordt een non-ketotische hyperglycinemie behandeld?
Geen genezing
Er bestaat geen behandeling die een non-ketotische hyperglycinemie kan genezen.
Kwaliteit van leven
Centraal in de behandeling van kinderen met deze aandoening staat het behouden van zo veel mogelijk kwaliteit van leven. Het is goed als ouders samen met hun andere kinderen en familie al in een vroeg stadium nadenken op wat voor manier zij hun kindje met deze aandoening willen begeleiden. Een maatschappelijk werkende, een psycholoog en verpleegkundigen van de kinderthuiszorg kunnen ouders hierbij helpen. In Nederland zijn ook meerdere kindercomfortteams die gezinnen kunnen ondersteunen bij het vinden van een zo optimaal mogelijke kwaliteit van leven.

Tijd voor samenzijn
De zorg voor een kindje met NKH zal veel vragen van ouders. Ouders zullen veel tijd kwijt zijn met verzorging van hun kind of zelfs met medische handelingen zoals het geven van medicijnen of sondevoeding. Het is ook heel belangrijk om er ook voor te waken dat er tijd blijft voor samen zijn als gezin of even als ouders onder elkaar, voor samen knuffelen waar kinderen met deze aandoening vaak van kunnen genieten en te zoeken naar manieren van contact waar iedereen plezier aan beleefd.
Hulp vragen aan anderen (bekenden of onbekenden) is voor veel ouders lastig, maar het is wel belangrijk om in een vroeg stadium na te denken over het vragen en organiseren van hulp. Dit om te voorkomen dat ouders de hele dag bezig zijn met zorgen en er geen tijd meer over is over fijne momenten samen met het kind met NKH de andere kinderen in het gezin en de ouders onderling.
Medicijn natriumbenzoaat
Het medicijn natriumbenzoaat 250-750 mg/kg/dag kan de hoeveelheid glycine in het bloed verminderen. Dit medicijn bindt aan glycine en zet het om in hippuurzuur wat via de nieren en de urine uit het lichaam wordt opgeruimd.
Wanneer dit medicijn gegeven wordt op een moment dat er relatief nog weinig klachten zijn, dan zou dit medicijn er voor kunnen zorgen dat kinderen zich beter ontwikkelen dan kinderen die dit medicijn niet krijgen. Een groot deel van de kinderen heeft al veel klachten op moment dat de diagnose gesteld wordt. Klachten die er al zijn, verbeteren vaak niet als gevolg van de behandeling met dit medicijn. Ook heeft dit medicijn nauwelijks effect op de hoeveelheid glycine in de hersenvloeistof, terwijl de kinderen juist het meest last hebben van symptomen die ontstaan in de hersenen. Natriumbenzoaat kan er voor zorgen dat de L-carnitine waarde in het bloed te laag wordt, waardoor het ook nodig is om L-carnitine te geven. Veel kinderen vinden de natriumbenzoaat vies smaken, wat kan maken dat het lastig is kinderen dit medicijn te geven.

Dextrometrofan
Naast natriumbenzoaat kan ook het medicijn dextrometrofan 5 mg/kg gegeven worden. Dit medicijn remt de overactieve NMDA receptor. Dit medicijn kan ook een bescheiden bijdrage leveren aan het verloop van deze ziekte.

Behandeling epilepsie
Er worden vaak verschillende medicijnen die epileptische aanvallen kunnen verminderen gegeven aan kinderen met NKH. Het is vaak erg moeilijk om een combinatie medicijnen te vinden die er voor zorgt dat kinderen geen epileptische aanvallen meer hebben. Meestal is het doel zo min mogelijk aanvallen met zo min mogelijk bijwerkingen als gevolg van het gebruik van medicijnen. Medicijnen die effect kunnen hebben op NKH zijn fenobarbital, clobazam, clonazepam en felbamaat. Het vrije nieuwe medicijn perampanel blijkt ook goed effect te kunnen hebben op epilepsie bij kinderen met NKH.
Een ketogeen dieet is een andere behandelmogelijkheid voor de epilepsie van kinderen met een NKH. Helaas is de epilepsie als gevolg van NKH zeer moeilijk behandelbaar.
In geval van een status epilepticus kan het anaestheticum ketamine worden ingezet. Ketamine remt ook de NMDA-receptor.

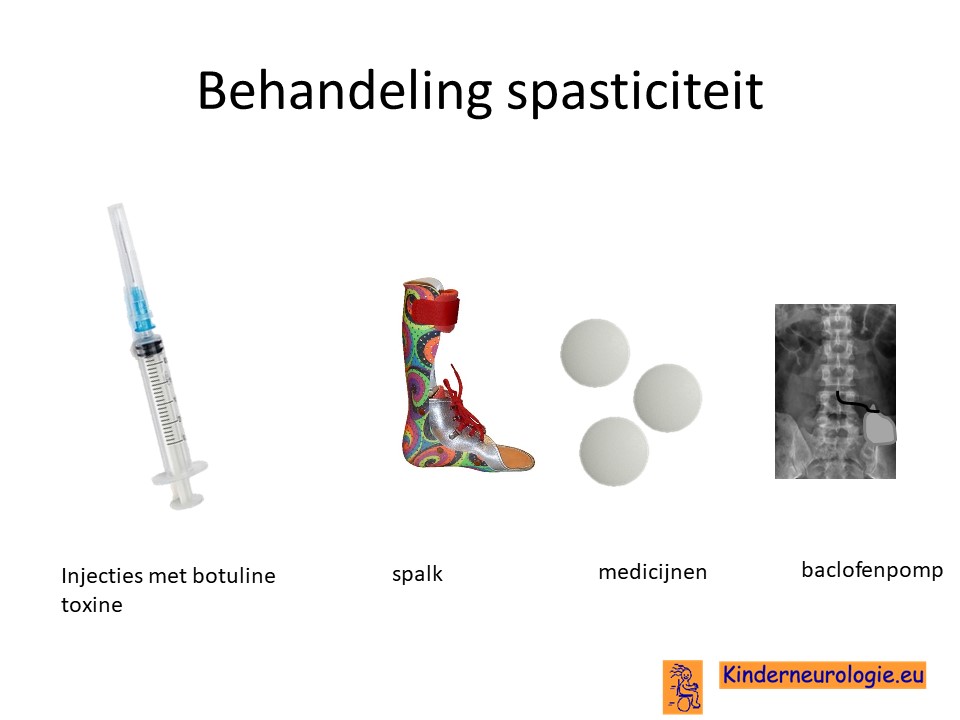
Behandeling spasticiteit
Injecties met botulinetoxine in een spastische spier kan de spasticiteit voor een aantal maanden verminderen. Met behulp van spalken en fysiotherapie kan op deze manier het looppatroon verbeterd worden. Vaak moeten deze injecties na een aantal maanden weer herhaald worden.
Ook kan met behulp van medicijnen geprobeerd worden om de spasticiteit van de benen te verminderen. Nadeel van al deze medicijnen is vaak dat ze de spierzwakte verergeren en in het hele lichaam effect hebben, niet alleen in de benen.
Veel gebruikte medicijnen zijn baclofen (Lioresal ®) en trihexyfenidyl (Artane®). Baclofen kan ook in de vorm van een baclofenpomp worden toegediend.
Bij ernstige spasticiteit kan het nodig zijn om met behulp van een operatie te zorgen dat kinderen minder last hebben van hun spasticiteit. Een veelvoorkomende operatie is het doornemen van de pezen van de spieren die er voor zorgen dat de bovenbenen strak tegen elkaar gedrukt worden. Dit belemmerd het lopen en de verzorging vaak ernstig. Na het doornemen van deze pezen verbeteren deze problemen vaak.
Behandeling dystonie
Er bestaan verschillende medicijnen die van invloed zijn op de dystonie en de dystonie kunnen verminderen. Het gaat dan om medicijnen als Baclofen (Lioresal®), Trihexyfenidyl (Artane®), clonazepam (Rivotril®) , L-Dopa of gabapentine (Neurontin®) of tetrabenazine (Tetmodis®). Er moet gezocht worden naar een juiste dosis van de medicijnen waarin er zoveel mogelijk effect is en er zo weinig mogelijk bijwerkingen zijn. Wanneer uw kind het lastig vindt om medicijnen in te nemen, vindt u hier tips voor innemen medicijnen. Baclofen kan ook door middel van een baclofenpomp worden toegediend.
Medicijnen die bij het ene kind wel werken, kunnen bij het andere kind geen effect hebben. Het blijft dus een kwestie van uitproberen wat het effect van een medicijn is en dit af te wegen tegen de bijwerkingen die het medicijn heeft.
Ook kunnen injecties met botuline toxine helpen wanneer de dystonie in een lichaamsdeel aanwezig is. Bij kinderen met een ernstige vorm van dystonie kan een deep brain stimulation worden overwogen. In Nederland wordt DBS bij kinderen toegepast in het Amsterdamumc en het UMC Groningen.

VISIO/Bartimeus
VISIO en Bartimeus zijn instellingen die kinderen en volwassenen die slechtziend of blind zijn begeleiden. Zij kunnen vaak tips hebben hoe kinderen die slecht kunnen zien het best kunnen spelen of benaderd kunnen worden.

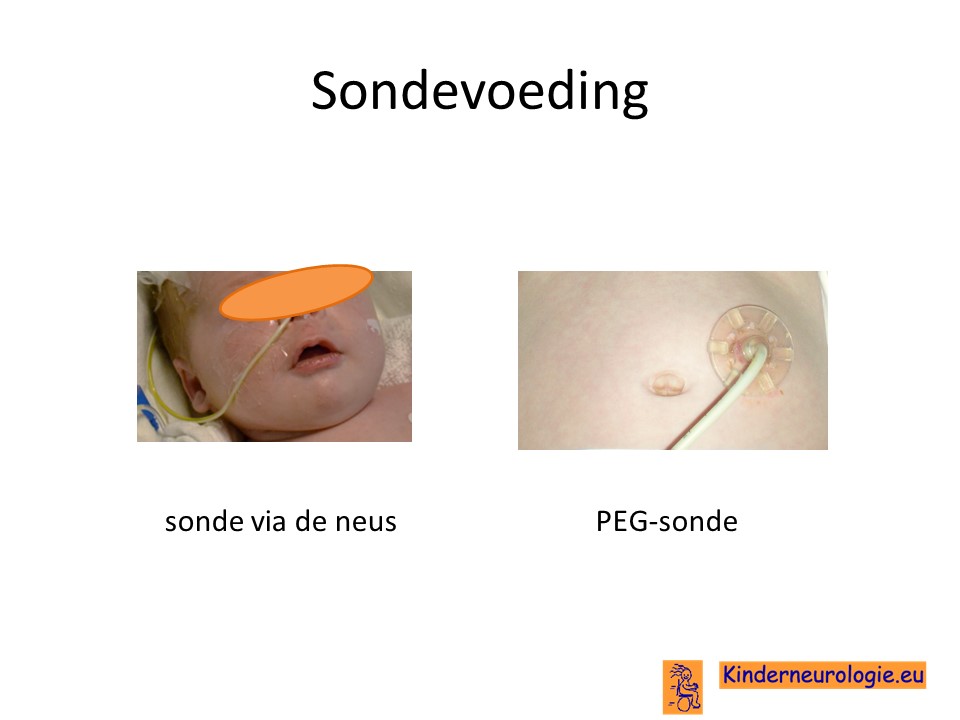
Sondevoeding
Een groot deel van de kinderen heeft niet voldoende kracht om zelf te eten en te drinken. Meestal krijgen kinderen daarom een sonde om op deze manier voldoende voeding en drinken binnen te krijgen. Wanneer kinderen langdurig een sonde nodig hebben, dan is het mogelijk om deze sonde rechtstreeks via de huid in de maag te laten eindigen. Dit wordt een PEG-sonde genoemd, later kan dit vervangen worden door een zogenaamde mickey button. Een deel van de ouders kiest er voor om fijn gepureerde voeding via de sonde te geven. Dit wordt een blended diet genoemd.
Kindercardioloog
Wanneer er sprake is van een verhoogde bloeddruk in de longen, kunnen medicijnen nodig zijn om de bloeddruk daar te verlagen.
Beademing
Baby’s met NKH hebben meestal beademing nodig om in leven te kunnen blijven. Beademing wordt gegeven op een intensive care afdeling voor pasgeborenen. Bij een deel van de kinderen kan de beademing na twee tot drie weken geleidelijk aan worden verminderd en daarna worden gestopt, bij een ander deel van de kinderen lukt dit niet. Beademing is een intensieve behandeling, het is goed dat de ouders en de dokters samen kijken of het kind deze intensieve behandeling te verantwoorden is naar het kind toe.
Fysiotherapie
Wanneer kinderen problemen krijgen met bewegen, kan een fysiotherapeut de spieren soepel houden door de benen en/of armen zelf te bewegen en tips te geven hoe een kind of de ouders dit zelf ook kunnen doen. Zo wordt voorkomen dat bepaalde gewrichten vast gaan groeien omdat ze te weinig bewogen worden. De fysiotherapeut kan tips geven om de ontwikkeling te stimuleren.
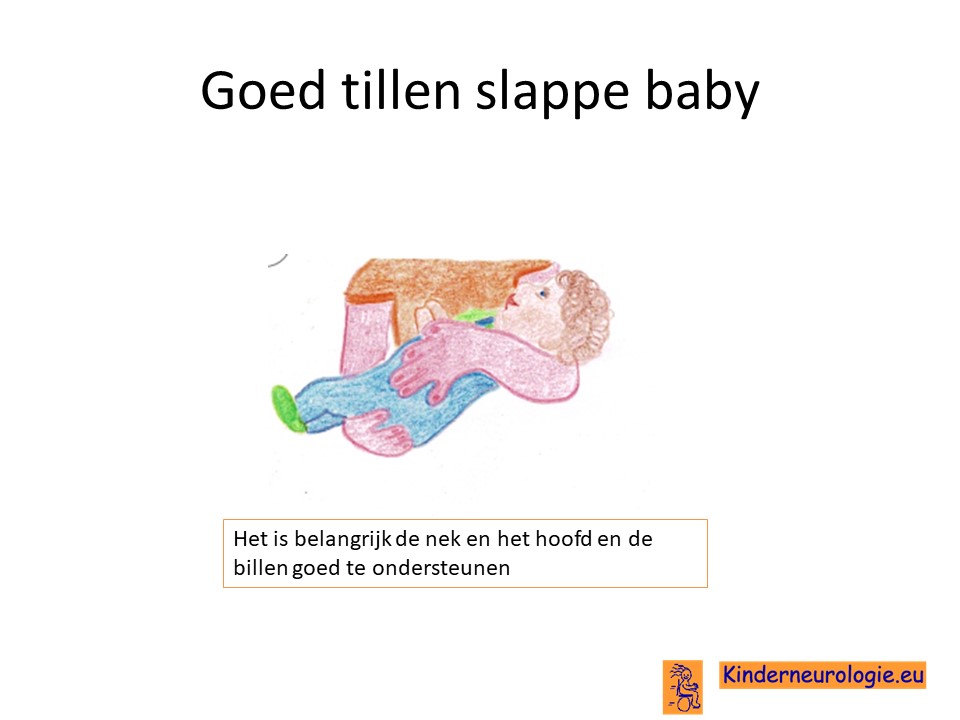
Logopedie
De logopedist kan adviezen geven hoe het slikken, eten en drinken zo goed mogelijk kan verlopen. Ook kan ze kinderen trainen in het goed en duidelijk praten of in het uitbreiden van de woordenschat. Wanneer praten moeilijk is, kan de logopediste andere manier van communiceren bijvoorbeeld met gebaren of met symbolen aanleren aan kind en ouders.

Ergotherapie
De ergotherapie kan advies geven over hulpmiddelen waarmee kinderen zo goed mogelijk verzorgd kunnen worden of waarmee kinderen zich zo goed mogelijk zelf kunnen redden.

Revalidatiearts
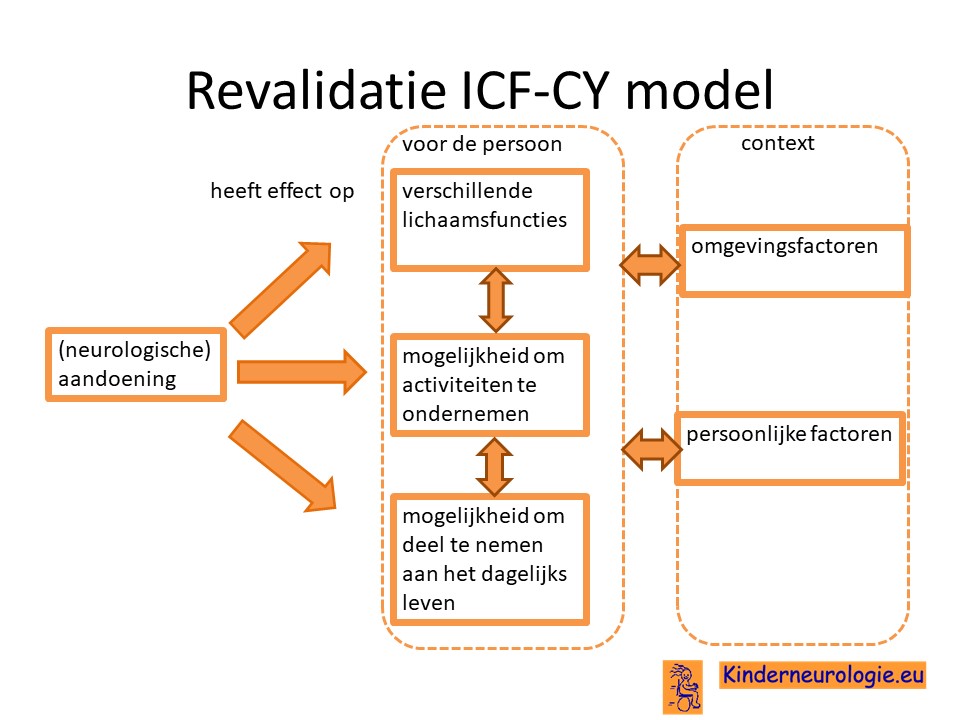
De revalidatiearts coördineert de verschillende behandelingen en vervolgt de ontwikkeling van kinderen met een ontwikkelingsachterstand. Bij problemen wordt gekeken wat voor oplossing er voor deze problemen te bedenken is. Vaak doet de revalidatiearts dit aan de hand van ICF-CY model. Er wordt gekeken wat het effect is van de aandoening op de verschillende lichaamsfuncties van het kind, de mogelijkheid om activiteit te ondernemen (bijvoorbeeld eten, aankleden, spelen) en de mogelijkheden om deel te nemen aan het dagelijks leven. De revalidatiearts denkt samen met een team mee welke oplossingen er te bedenken zijn voor een bepaald probleem.
De revalidatiearts geeft ook adviezen voor aangepaste schoenen of het gebruik van bijvoorbeeld spalken.
De revalidatiearts geeft ook adviezen voor het juiste onderwijs van het kind. Sommige kinderen gaan naar de school die verbonden is aan het revalidatiecentrum.
Ook voor jongere kinderen bestaan op het revalidatiecentrum vaak groepjes waarin de kinderen gedurende een dagdeel therapie krijgen waarin hun ontwikkeling gestimuleerd wordt.
Dagopvang
Vanaf de leeftijd van 2 maanden kunnen kinderen die niet naar een reguliere kinderdagopvang kunnen, naar een speciale kinderdagopvang toe gaan. Er bestaat speciale therapeutische peutergroepen in revalidatiecentra, of dagopvang in een orthopedagogisch dagcentrum (ODC) of in een medische kinderdagcentrum (MKD). Het hangt van de problemen die het kind ervaart af (zoals epilepsie of gedragsproblemen), welke vorm van dagopvang het meest geschikt is. Aanmelding voor een ODC of een MKD verloopt via de gemeente (vaak cia het centrum jeugd en gezin, via het jeugdteam of via het sociaal wijkteam). Aanmelding voor een therapeutische peutergroep in een revalidatiecentrum verloopt via de revalidatiearts.

School
De meeste kinderen met NKH volgen speciaal onderwijs. Hier zijn de klassen kleiner en kan het lesprogramma meer afgestemd worden op de mogelijkheden van het kind. Vaak volgen kinderen MLK (moeilijk lerend) of ZMLK (zeer moeilijk lerend) onderwijs.
Voor een deel van de kinderen is het niet haalbaar om onderwijs te volgen. Zij gaan naar een dagcentrum waar kinderen een dagprogramma volgen.Het LWOE kan leerkrachten adviezen geven hoe kinderen met epilepsie op school het beste begeleid kunnen worden.
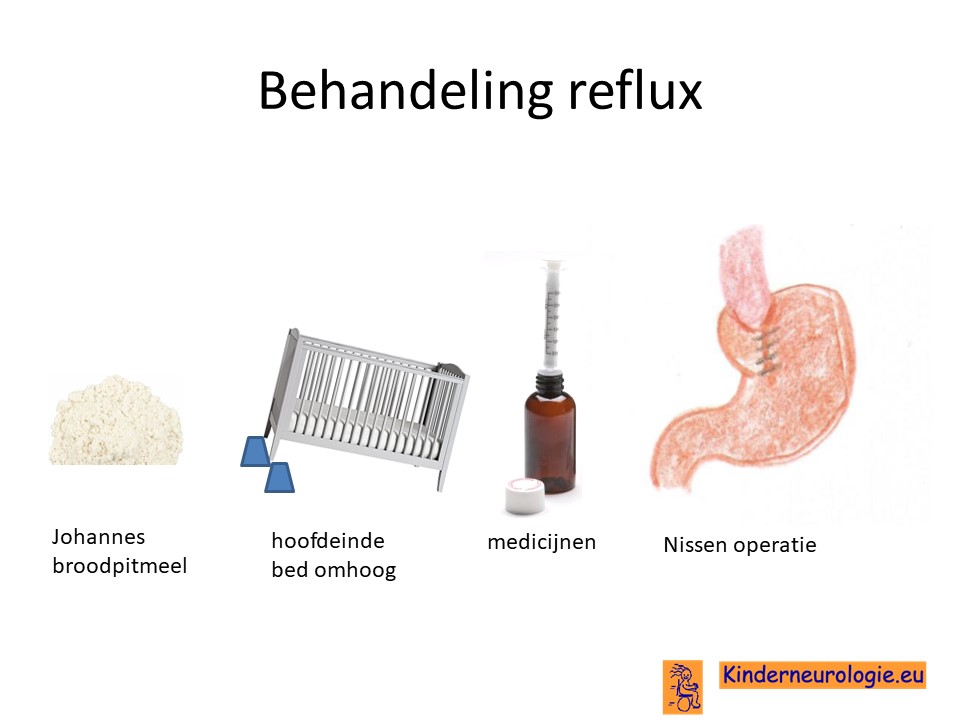
Reflux
Reflux kan er ook voor zorgen dat kinderen slecht eten. Door de voeding in te dikken met johannesbroodpitmeel kan de voeding minder gemakkelijk terug stromen van de maag naar de slokdarm. Ook zijn er medicijnen die de maaginhoud minder zuur kunnen maken waardoor de slokdarm minder geprikkeld wordt bij terugstromen van de maaginhoud. Medicijnen die hiervoor gebruikt worden zijn ranitidine en omeprazol, soms esomeprazol. Indien dit allemaal niet voldoende is, kan een operatie nodig zijn waarbij de overgang van de slokdarm naar de maag nauwer wordt gemaakt, waardoor de voeding ook minder gemakkelijk terug kan stromen. Dit wordt een Nissen-operatie genoemd.
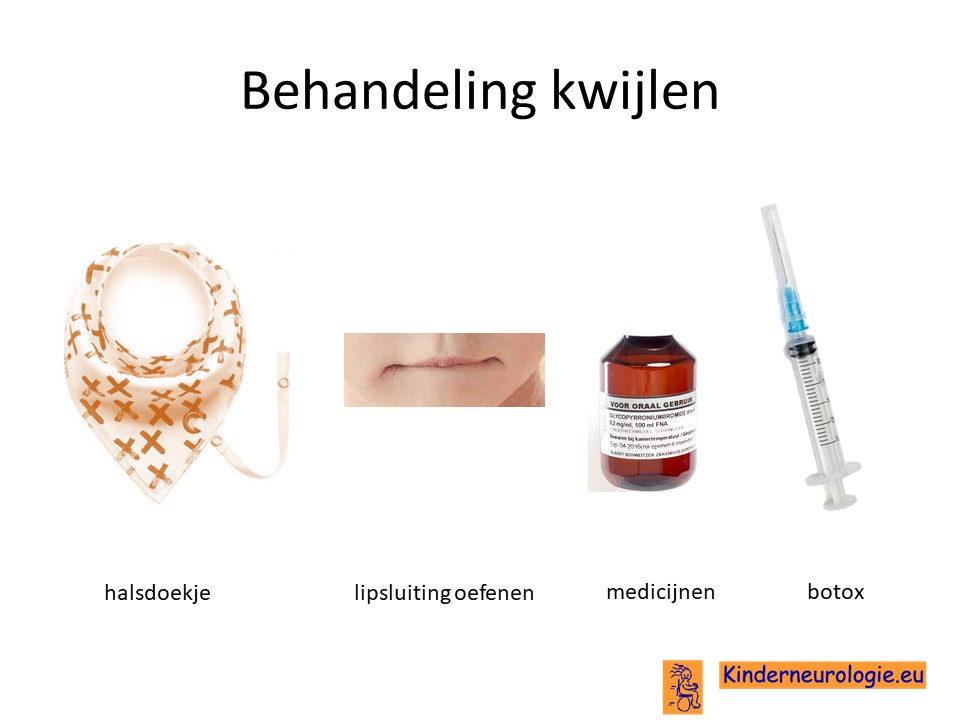
Kwijlen
Kwijlen kan verminderen door kinderen er bewust van te maken dat ze hun speeksel moeten doorslikken. Ook kunnen oefeningen waarbij geoefend wordt om de mond te sluiten helpen. Er bestaan moderne halsdoekjes die kwijl kunnen opvangen, zodat de kleding niet vies en nat wordt.
Er bestaan medicijnen die het kwijlen minder kunnen maken. Het meest gebruikte medicijn hierdoor is glycopyrrhonium. Soms kan een behandeling van de speekselklieren door middel van botox of door middel van een operatie nodig zijn om er voor zorgen dat kinderen minder kwijlen. Per kind zullen de voor- en nadelen van elke behandeling moeten worden afgewogen.
Verstopping van de darmen
Het medicijn macrogol kan er voor zorgen dat de ontlasting soepel en zacht blijft en stimuleert de darmwand om actief te blijven. Hierdoor kunnen kinderen gemakkelijker hun ontlasting kwijt. Verder blijft het belangrijk om te zorgen dat kinderen voldoende vocht en vezels binnen krijgen en zo veel als kan bewegen. Soms zijn zetpillen nodig om de ontlasting op gang te krijgen.

Zindelijkheid
Er kan met zindelijkheidstraining worden begonnen wanneer het kind zelf kan zitten op een potje en interesse begint te krijgen in het potje. Vaak is dit bij kinderen met dit syndroom op latere leeftijd dan gebruikelijk. Tips die kunnen helpen bij het zindelijk worden vindt u in de folder zindelijkheid. Voor een groot deel van de kinderen met de klassieke vorm van NKH is het niet haalbaar om zindelijk te worden.
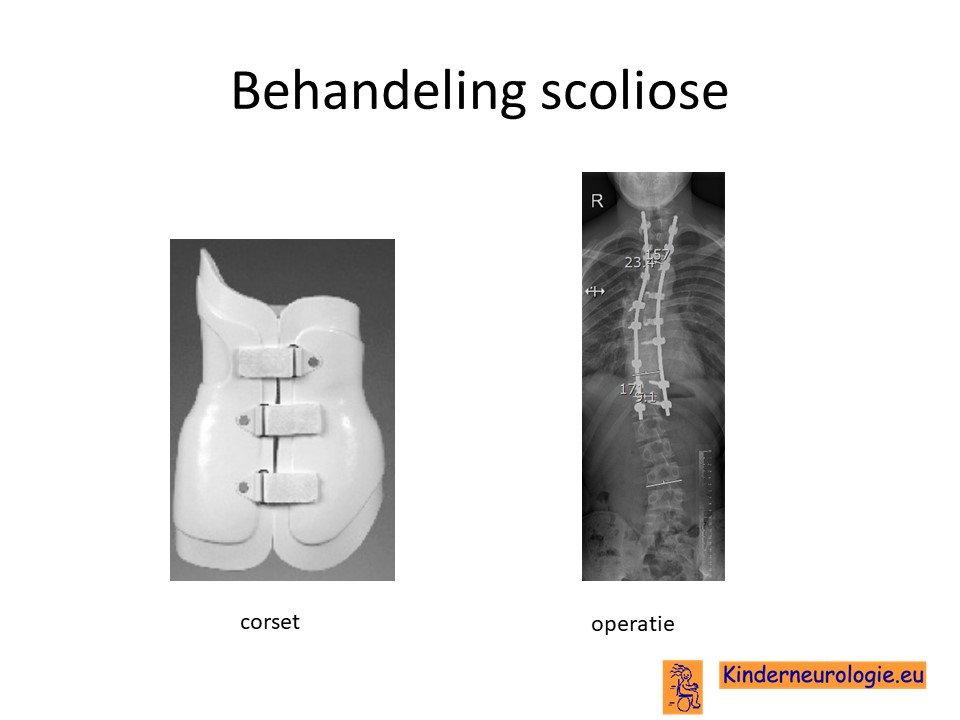
Scoliose
Lichte vormen van verkromming van de wervelkolom hebben meestal geen behandeling nodig en alleen controle om te kijken of de verkromming toeneemt. Bij toename kan een behandeling met een gipscorset nodig zijn om verdergaande verkromming van de wervelkolom te voorkomen. Wanneer een gipscorset onvoldoende effect heeft, kan een operatie nodig zijn waarbij de wervels vastgezet. Deze behandeling wordt uitgevoerd door een orthopeed.
Waterhoofd
Een waterhoofd kan behandeld worden door de neurochirurg. Vaak is een drain nodig, een slangetje die overtollig vocht uit de hersenkamers afvoert naar de buikholte of het hart.
Financiële kant van zorg voor een kind met een beperking
De zorg voor een kind met een beperking brengt vaak extra kosten met zich mee. Er bestaan verschillende wetten die zorg voor kinderen met een beperking vergoeden.
Daarnaast bestaan regelingen waar ouders een beroep op kunnen doen, om een tegemoetkoming te krijgen voor deze extra kosten. Meer informatie hierover vindt u in de folder financiën kind met een beperking.

Kindercomfortteam
In Nederland zijn in de academische ziekenhuizen speciale kindercomfortteams. Dit zijn teams bestaande uit meerdere hulpverleners (verpleegkundigen, pedagogisch medewerkers, maatschappelijk werkenden, psychologen, geestelijke verzorgging, artsen) die ervaring hebben met de zorg voor kinderen met een aandoening die niet te genezen is. Dit team kijkt samen met kind en ouders hoe het kind een zo goed mogelijke kwaliteit van leven kan krijgen en hoe kind en ouders hierin zo goed mogelijk ondersteund kunnen worden. Dit kan per kind en ouders verschillen.

Wat kun je als ouder zelf doen om de ontwikkeling van je kind optimaal te laten verlopen?
Bedenk dat wanneer je samen met je kind speelt, stoeit, danst, zingt, kletst, lacht en/of boekjes leest, dit ook allemaal manieren zijn waarop je kind zijn of haar hersenen traint om stappen voorwaarts te maken in de ontwikkeling. Het is dus niet zo dat alleen momenten van therapie, momenten van training zijn, wat veel ouders denken. Het is daarnaast goed om inspanning af te wisselen met ontspanning, dit is nodig om het geleerde te laten opslaan in de hersenen. De hele dag door training zonder rustmomenten, werkt juist averechts.
Daarnaast is het van onschatbare waarde je kind laten voelen dat je van hem of haar houdt, dat hij/zij geliefd is en zich mag ontwikkelen in een tempo die bij hem of haar past. Dit is extra van belang voor kinderen die zich anders ontwikkelen dan de "norm". "Goed zijn zoals je bent en gesteund te worden door mensen die van je houden is, heel belangrijk voor de ontwikkeling van een kind. Juist de ouders en de andere kinderen in het gezin die dichtbij het kind staan zijn daarin heel belangrijk om het kind daarin dit gevoel te geven. Het is goed dat ouders beseffen wat de waarde hiervan is voor het kind en welke rol zij hierin hebben.
Ook is het belangrijk om te bedenken wat goed voelt voor jullie als gezin en voor jou als ouder en waar jullie energie uithalen. Zorg ervoor dat er bewust ruimte is voor momenten die dit goede gevoel geven. Tot slot is het belangrijk dat je als ouders ook goed voor jezelf zorgt, de zorg voor een kind die zich anders ontwikkelt vraagt nog meer van ouders dan de zorg voor een kind die zich zonder problemen ontwikkelt. Het is goed om voor jezelf te zorgen of te laten zorgen, zodat je als ouder ook de energie houdt, om jouw kind te blijven begeleiden op een manier die bij jou past. Besef dat bij opvoeden hoort om te leren los laten. Veel ouders vinden dit lastig, zeker wanneer hun kind zich anders ontwikkelt dan andere kinderen. Maar dhet kan toch nodig zijn een deel van de zorg op bepaalde momenten uit handen te geven, ook als die ander het anders doet dan jij, je kind leert van deze verschillen en het geeft jou de mogelijk om zelf uit te rusten of nieuwe energie op te doen.

Wat kun je als gezin zelf doen om om te gaan met het hebben van een aandoening bij een gezinslid?
Als gezin van een kind waarbij er sprake is van een aandoening, is het goed om te zorgen dat jullie in de je kracht komen staan. Het is goed om te beseffen over welke denk-, emotionele-, innerlijke- en fysieke kracht jullie als gezin beschikken en hoe jullie deze kracht kunnen inzetten om goed voor ieder lid van het gezin te zorgen. Bekijk wat bij jullie als gezin past. Bekijk wat je kunt doen (of kunt laten) om deze kracht zo optimaal mogelijk in te zetten. En bedenk ook dat ieder lid van het gezin verschillende kwaliteiten heeft waarmee jullie elkaar kunnen aanvullen en kunnen versterken.
Begeleiding
Begeleiding van kinderen met NKH en hun ouders is belangrijk. Een maatschappelijk werkende of een psycholoog kunnen kind en ouders begeleiden in het omgaan met het hebben van deze ziekte en met de gevolgen van deze ziekte.Ook vinden veel ouders het vaak lastig hoe zij hun tijd en aandacht moeten verdelen tussen het kind met de beperking en andere kinderen in het gezin. In de folder aandacht en tijd voor brussen vindt u tips die u hierbij kunnen helpen.

Contact met andere ouder
Door het plaatsen van een oproepje op het forum van deze site kunt in contact komen met andere kinderen met NKH en hun ouders of verzorgers.
Wat betekent heb hebben van een non-ketotische hyperglycinemie voor de toekomst?
Ontwikkelingsacherstand
De meeste kinderen met een non-ketotische hyperglycinemie ontwikkelen zich heel langzaam.
Kinderen die op vroege leeftijd de eerste symptomen van NKH hebben gekregen, zijn veel verder achter in hun ontwikkeling dan kinderen die pas op latere leeftijd symptomen hebben gekregen.
Moeilijk behandelbare vorm van epilepsie
Veel kinderen met een non-ketotische hyperglycinemie hebben een moeilijk behandelbare vorm van epilepsie. Vaak lukt het niet om de epilepsie met behulp van medicijnen of een ketogeen dieet helemaal onder controle te krijgen. Doel van de behandeling is dan om er voor te zorgen dat kinderen zo min mogelijk last hebben van hun aanvallen, zonder dat ze al te veel bijwerkingen hebben als het gevolg van de medicijnen die nodig zijn voor de behandeling van de epilepsie.
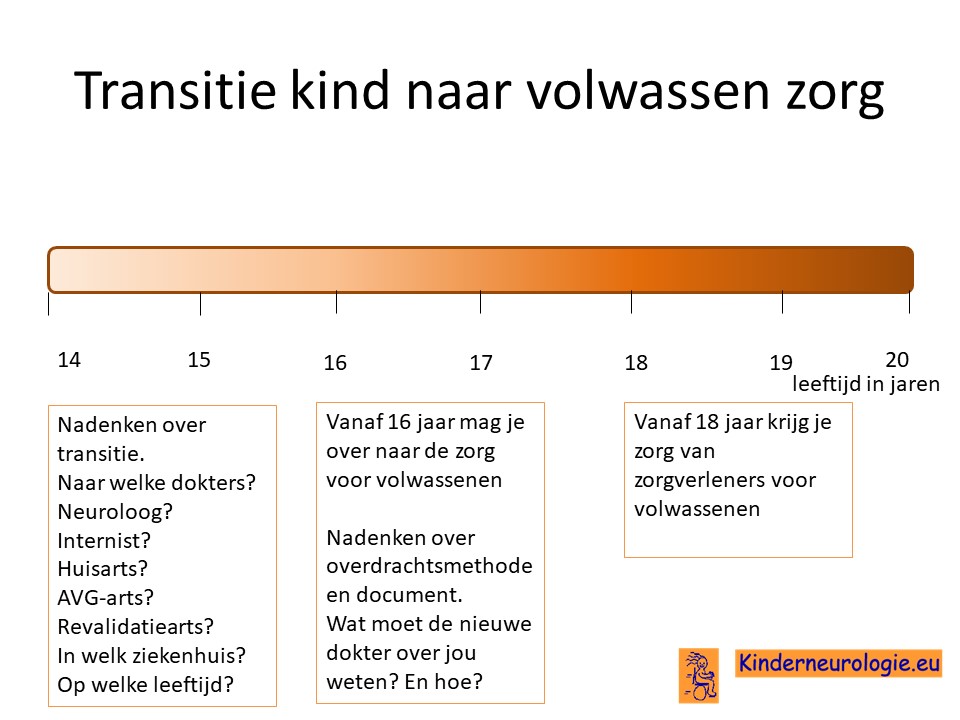
Transitie van zorg
Tussen de leeftijd van 16 en 18 jaar wordt de zorg vaak overgedragen van kinderspecialisten naar specialisten die de zorg aan volwassenen geven. Het is belangrijk om tijdig hierover na te denken. Is er behoefte de zorg over te dragen naar specialisten voor volwassenen of kan de huisarts de zorg leveren die nodig is.En als er behoefte is aan overdragen van de zorg naar specialisten voor volwassenen, naar welke dokter(s) wordt de zorg dan overgedragen? In welk ziekenhuis kan de zorg het beste geleverd worden. Het proces van overdragen van de zorg wordt transitie genoemd. Het is belanrgijk hier tijdig over na te denken en een plan voor te maken samen met de dokters die betrokken zijn bij de zorg op de kinderleeftijd. Ook verandert er veel in de zorg wanneer een jongere de leeftijd van 18 jaar bereikt. Voor meer informatie over deze veranderingen verwijzing wij u naar het artikel veranderingen in de zorg 18+
Arts VG
Een Arts VG is een arts die zich gespecialiseerd heeft in de zorg voor mensen met een verstandelijke beperking. De Arts VG richt zich op het voorkomen, behandelen en beperken van lichamelijke en psychische problemen die te maken hebben met een verstandelijke of lichamelijke beperking. De Arts VG werkt hiervoor samen met de huisarts, de medische specialist, de gedragsdeskundige en/of andere therapeuten (zoals een fysiotherapeut of een logopedist). Er zijn steeds meer poliklinieken in Nederland waar een Arts VG werken en waar kinderen en volwassenen met een verstandelijke beperking terecht kunnen met hun hulpvragen die te maken hebben met hun beperking. Daarnaast werken Artsen VG ook in instellingen en zijn ze betrokken bij gespecialiseerde kinderdagcentra. Op de website van de VGN is een lijst met poliklinieken te vinden waar Artsen VG werken.

Levensverwachting
Het hebben van non-ketotische hyperglycinemie heeft invloed op de levensverwachting. Vooral de problemen met de ademhaling kunnen levensbedreigend zijn.
Een deel van de kinderen komt helaas op jonge leeftijd te overlijden.
Kinderen krijgen
De meeste kinderen met een non-ketotische hyperglycinemie zijn dusdanig beperkt in hun ontwikkelingsmogelijkheden dat zij zelf geen kinderen zullen krijgen. De kans dat een volwassene met NKH zelf een kind met NKH gaat krijgen is heel klein, dit kan alleen wanneer de partner drager is van een zelfde fout in het DNA of wanneer de partner zelf NKH heeft. De kans hierop is erg klein.
Verbetering van de symptomen
Bij een heel klein deel van de kinderen nemen de klachten weer af in de loop van de maanden. Op een of andere manier is het lichaam weer in staat om glycine op te ruimen. De ontwikkeling gaat weer verder en kinderen hebben niet meer zo veel last van epilepsie aanvallen. Vaak zijn er wel milde leerproblemen en gedragsproblemen, maar deze kinderen zijn wel weer in staat om redelijk zelfstandig deel te nemen aan de maatschappij. Het is niet bekend wat maakt dat glycine waardes bij deze kinderen weer dalen. Jongens blijken een grotere kans te hebben op deze stabilisatie dan meisjes, de reden hiervan is niet bekend.
Hebben broertjes en zusjes een vergrote kans om ook non-ketotische hyperglycinemie te krijgen?
Non-ketotische hyperglycinemie is een ziekte die veroorzaakt wordt door een foutje in het erfelijk materiaal. De ziekte erft op zogenaamd autosomaal recessieve wijze over. Vaak zijn beide ouders drager van de ziekte. Zij hebben een chromosoom met een fout en een chromosoom zonder fout, zodat zij zelf geen klachten hebben. Hun kinderen hebben 25 % kans om ook non-ketotische hyperglycinemie te krijgen. Een klinisch geneticus kan hier meer informatie over geven.
Familiebrief
Het hebben van een genetische aandoening kan soms ook consequenties hebben voor andere familieleden dan alleen de jongere en zijn gezin. Er kan een kans bestaan dat deze aandoening bij meerdere familieleden voorkomt. Een klinisch geneticus maakt meestal een familiebrief. Hierin wordt uitgelegd wat de aandoening inhoudt, waar meer informatie te vinden is over de aandoening en waarin vermeld staat of familieleden een verhoogde kans hebben om ook zelf deze aandoening te hebben. Met deze brief kunnen familieleden die dat willen via de huisarts verwezen worden naar een klinisch geneticus.
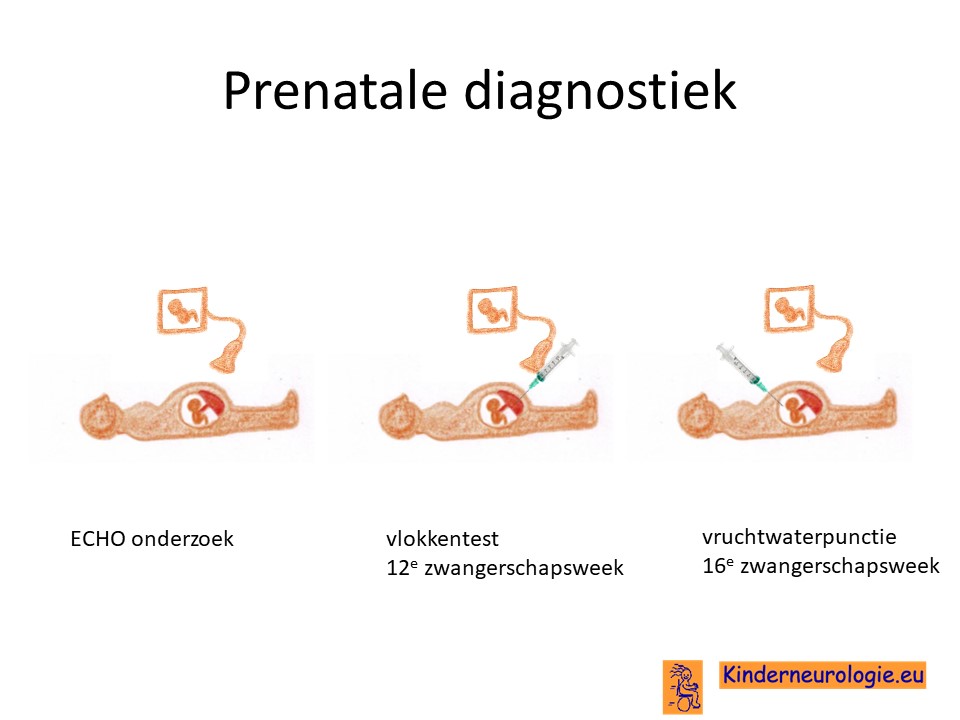
Prenatale diagnostiek
Wanneer bekend is welk foutje in het erfelijk materiaal de non-ketotische hyperglycinemie veroorzaakt heeft, dan bestaat er tijdens een zwangerschap van een broertje of zusje de mogelijkheid om een prenatale diagnostiek uit te voeren. Door middel van een vruchtwaterpunctie wordt erfelijk materiaal verzameld, zodat gekeken kan worden of er in dit materiaal ook foutjes zitten genen die non-ketotische hyperglycinemie veroorzaken. Beide ingrepen hebben een klein risico op het ontstaan van een miskraam (0,5% bij de vlokkentest en 0,3% bij de vruchtwaterpunctie).De uitslag van deze onderzoeken duurt twee weken. Voor prenatale diagnostiek kan een zwangere de 8ste week verwezen worden door de huisarts of verloskundige naar een afdeling klinische genetica. Meer informatie over prenatale diagnostiek kunt u vinden op de website: www.pns.nl
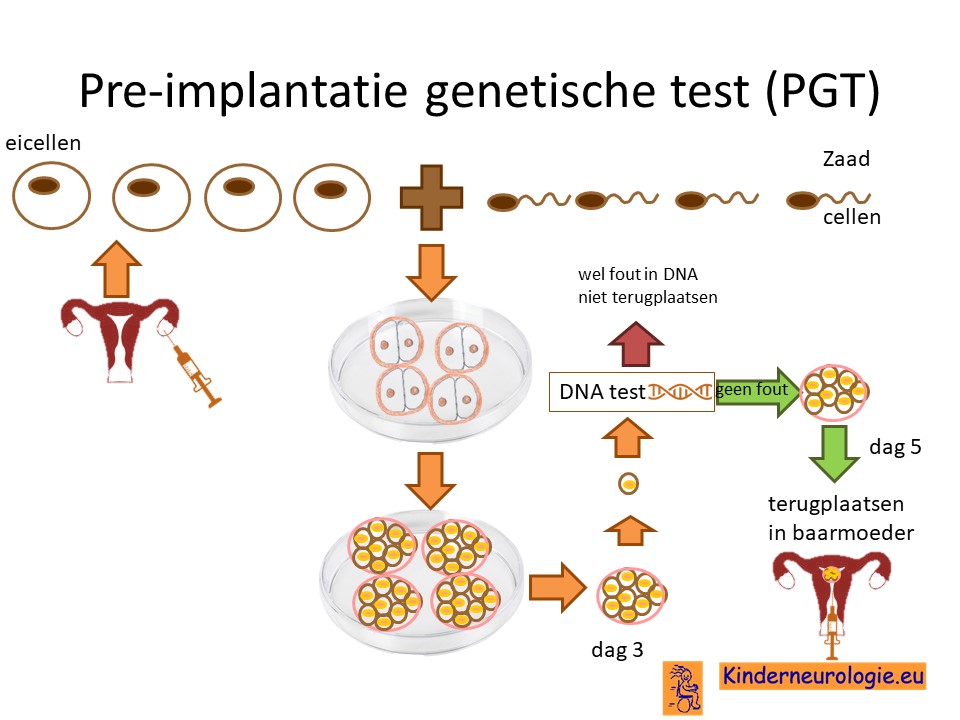
Preïmplantatie Genetische Test (PGT)
Stellen die eerder een kindje hebben gehad met non-ketotische hyperglycinemie kunnen naast prenatale diagnostiek ook in aanmerking voor een preïmplantatie genetische test (PGT.) Bij PGT wordt een vrouw zwanger door middel van IVF (In Vitro Fertilisatie). De bevruchting vindt dan buiten het lichaam plaats, waardoor het zo ontstane pre-embryo onderzocht kan worden op het hebben van non-ketotische hyperglycinemie. Alleen embryo’s zonder de aanleg voor non-ketotische hyperglycinemie komen in aanmerking voor terugplaatsing in de baarmoeder. Voor meer informatie zie www.pgtnederland.nl.
Wilt u dit document printen dan kunt u hier een pdf-versie downloaden.
Wilt u ook uw verhaal kwijt, dat kan: verhalen kunnen gemaild worden via info@kinderneurologie.eu en zullen daarna zo spoedig mogelijk op de site worden geplaatst. Voor meer informatie zie hier.
Heeft u foto's die bepaalde kenmerken van deze aandoening duidelijk maken en die hier op de website mogen worden geplaatst, dan vernemen wij dit graag.
Links
www.stofwisselingsziekten.nl
(Nederlandse vereniging van mensen met een stofwisselingsziekte)
www.2cu.nu
(stichting complex care united, een stichting die zich inzet om gezinnen met een zeer ernstig verstandelijk en meervoudig beperkt kind een betere kwaliteit van leven te geven middels concrete hulp en ondersteuning)
Referenties
1. A known and a novel mutation in the glycine decarboxylase gene in a newborn with classic nonketotic hyperglycinemia. Beijer P, Lichtenbelt KD, Hofstede FC, Nikkels PG, Lemmers P, de Vries LS. Neuropediatrics. 2012;43:164-7
2. Two novel missense mutations observed in nonketotic hyperglycinemia. Yoon IA, Lee NM, Yoo BH, Lee BS, Yoo HW. Pediatr Neurol. 2012;46:401-3
3. Ketogenic diet in early myoclonic encephalopathy due to non ketotic hyperglycinemia. Cusmai R, Martinelli D, Moavero R, Dionisi Vici C, Vigevano F, Castana C, Elia M, Bernabei S, Bevivino E. Eur J Paediatr Neurol. 2012;16:509-13
4. Prediction of long-term outcome in glycine encephalopathy: a clinical survey. Hennermann JB, Berger JM, Grieben U, Scharer G, Van Hove JL. J Inherit Metab Dis. 2012;35:253-61
5. Nonketotic hyperglycinemia presenting as pulmonary hypertensive vascular disease and fatal pulmonary edema in response to pulmonary vasodilator therapy. Menéndez Suso JJ, Del Cerro Marín MJ, Dorao Martínez-Romillo P, Labrandero de Lera C, Fernández García-Moya L, Rodríguez González JI. J Pediatr. 2012;161:557-9
6. Nonketotic Hyperglycinemia: Two Case Reports and Review. Poothrikovil RP, Al Thihli K, Al Futaisi A, Al Murshidi F. Neurodiagn J. 2019;59:142-151
7. Use of Perampanel and a Ketogenic Diet in Nonketotic Hyperglycinemia: A Case Report. Daida A, Hamano SI, Ikemoto S, Hirata Y, Matsuura R, Koichihara R, Oba D, Ohashi H. Neuropediatrics. 2020;51:417-420
8. Metabolic epilepsies amenable to ketogenic therapies: Indications, contraindications, and underlying mechanisms. Gavrilovici C, Rho JM. J Inherit Metab Dis. 2021;44:42-53
9. Cerebrospinal fluid amino acids glycine, serine, and threonine in nonketotic hyperglycinemia. Swanson MA, Miller K, Young SP, Tong S, Ghaloul-Gonzalez L, Neira-Fresneda J, Schlichting L, Peck C, Gabel L, Friederich MW, Van Hove JLK. J Inherit Metab Dis. 2022;45:734-747
10. Integrative Approach to Predict Severity in Nonketotic Hyperglycinemia. Hübschmann OK, Juliá-Palacios NA, Olivella M, Guder P, Zafeiriou DI, Horvath G, Kulhánek J, Pearson TS, Kuster A, Cortès-Saladelafont E, Ibáñez S, García-Jiménez MC, Honzík T, Santer R, Jeltsch K, Garbade SF, Hoffmann GF, Opladen T, García-Cazorla Á. Ann Neurol. 2022;92:292-303
11. Glycine disrupts myelin, glutamatergic neurotransmission, and redox homeostasis in a neonatal model for non ketotic hyperglycinemia. Parmeggiani B, Signori MF, Cecatto C, Frusciante MR, Marcuzzo MB, Souza DG, Ribeiro RT, Seminotti B, Gomes de Souza DO, Ribeiro CAJ, Wajner M, Leipnitz G. Biochimie. 2023:S0300-908400183-9
Auteur: J.H. Schieving
Laatst bijgewerkt: 23 augustus 2023 voorheen: 27 juli 2022, 23 juni 2021, 28 oktober 2020, 20 januari 2020, 22 maart 2019 en 11 december 2012
Heeft uw kind nog andere symptomen, laat het ons weten.
