
Segmentale spinale spieratrofie

ALS is een neurodegeneratieve aandoening. Hierbij is niet alleen de voorhoorncel aangedaan, maar ook de pyramidebaan. Het is dus een aandoening van zowel het perifere als het centrale zenuwstelsel

Epidemiologie
ALS komt vooral op middelbare leeftijd voor, vaker bij mannen dan bij vrouwen. Bij een op de tien patienten is er sprake van een familaire vorm van ALS.

Pathofysiologie
De pathofysiologie van ALS is deels nog onopgehelderd. Waarschijnlijk is er sprake van een cascade van meerdere stappen (geschat 6 stappen) die er voor zorgt dat ALS ontstaat en motore voorhoorncellen en neuronen in de hersenen progressief verloren gaan.

Er zijn een aantal risicofactoren bekend die een bijdrage kunnen spelen bij het ontstaan van ALS.

Bepaalde veranderingen in het DNA maken mensen kwetsbaarder voor het ontwikkelen van ALS. Alleen het hebben van een fout in het DNA is onvoldoende om ALS te krijgen, er zijn ook nog andere factoren nodig die maken dat er daadwerkelijk ALS ontstaat. Gemiddeld zijn nog 4 stappen (in plaats van 6 bij mensen zonder fout in het DNA) nodig om ALS te doen ontstaan.

Symptomen ALS
Kenmerkend voor ALS zijn motore symptomen. Sensibele symptomen horen niet bij ALS !! De symptomen zijn langzaam progressief.
Fasciculaties komen vaak voor bij ALS, maar zijn er zeker niet specifiek voor. Fasciculaties in de kuiten komen bij 40-70% van de gezonde mensen voor. Fasciculaties op andere plaatsen komen ook bij andere neuromusculaire aandoeningen voor.

De eerste klachten kunnen ontstaan in een arm, in het gezicht (bulbair, met uitzondering van de oogspieren) of in een been. De klachten kunnen aan twee kanten van het lichaam aanwezig zijn, maar zijn vaak asymmetrisch. Dit maakt dat ALS een zeer heterogene aandoening is, die zich op allerlei manieren kan presenteren. Bij een klein deel van de patienten zijn respiratoire insufficientie of fors gewichtverlies de eerste symptomen van ALS.

ALS is een progressieve aandoening.

ALS is niet uitsluitend een motore aandoening, er komen ook cognitieve en gedragsmatige symptomen voor. Bij 5-15% van de mensen kan ook de diagnose frontotemporale dementie gesteld worden. De andere patienten voldoen niet aan de criteria voor FTD, bij hen wordt er gesproken van ALS-ci als de cognitieve symptomen op de voorgrond staan, van ALS-bi als de gedragsproblemen op de voorgrond staan en van ALS-cbi als er zowel cognitieve als gedragsproblemen zijn. Deze cognitieve en gedragsmatige symptomen hebben een grote impact op de kwaliteit van leven.

De diagnostiek bestaat uit het aantonen van stoornis van zowel het centrale als het perifere zenuwstelsel en het uitsluiten van andere aandoeningen die veel op ALS kunnen lijken.


Er bestaat een familaire vorm van ALS. Hierbij lukt het om bij 2/3 van de patienten een fout in het DNA aan te tonen die verantwoordelijk is voor het ontstaan van ALS. Maar ook bij de niet familaire vorm wordt bij een deel van de patienten (10%) een fout in het DNA gevonden.

Het blijkt dat mutaties in deze genen zeer verschillende ziektebeelden kunnen veroorzaken. Het is van belang om bij de familieanamnese niet alleen te vragen naar het voorkomen van ALS in de familie, maar ook naar andere neurologische en psychiatrische aandoeningen.

Het is belangrijk om ALS-mimics uit te sluiten.
Sensibele symptomen, autonomen symptomen en oogbewegingsstoornissen horen niet bij ALS en betekenen dat er gezocht moet worden naar een andere diagnose. Dit zijn rode vlaggen voor de diagnose ALS.
Bij oranje vlaggen is de diagnose ALS wel mogelijk, maar is het verstandig om te zoeken of er toch geen sprake is van een andere aandoening.


Een deel van de diagnostiek is gericht op het uitsluiten van deze mimics.

Behandeling ALS
ALS is helaas niet te genezen. Met het medicijn riluzole kan het ziektebeloop met enkele maanden vertraagd worden. Ook kan een magistrale bereiding van dextromethorfan met kinidine zorgen voor een tijdelijke verbetering van bulbaire symptomen.

Het is belangrijk dat patienten met ALS behandeld worden door een multidisciplinaire team met diverse professionals met ervaring met ALS. Er bestaan in Nederland meerdere ALS zorgnetwerken waar patienten met ALS behandeld kunnen worden.

De behandeling van ALS is symptomatisch. vaak bestaan er meerdere behandelopties en bekijken de persoon met ALS en de behandelaar welke behandeling het beste past in de specifieke situatie van de persoon met ALS.
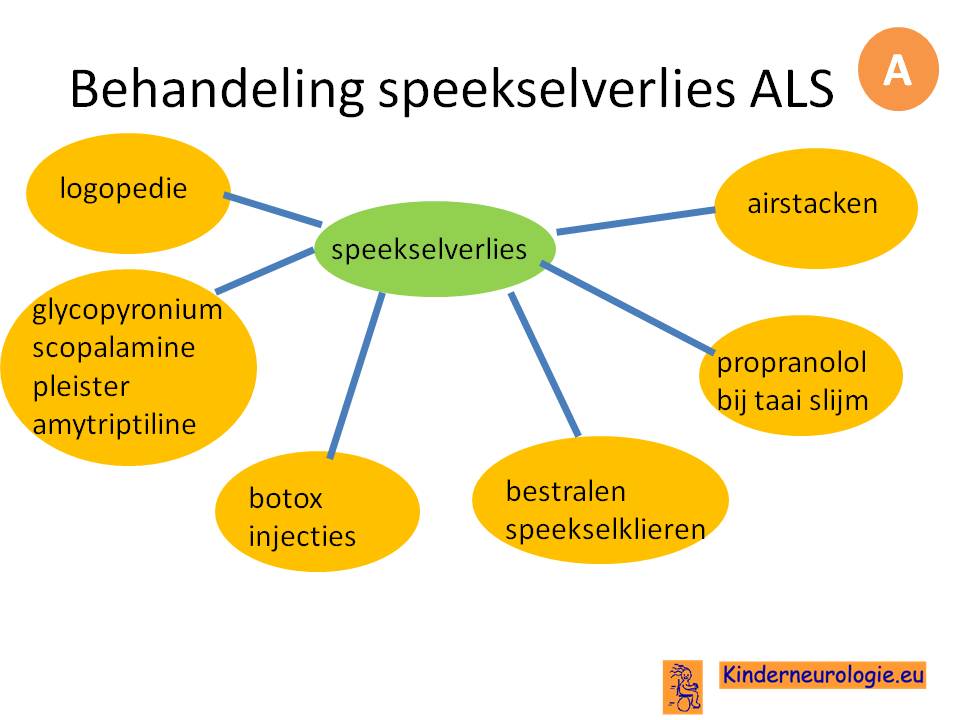

Het optimaliseren van de voedingstoestand en het voorkomen van gewichtsverlies zijn erg belangrijk voor de kwaliteit van leven van mensen met ALS.


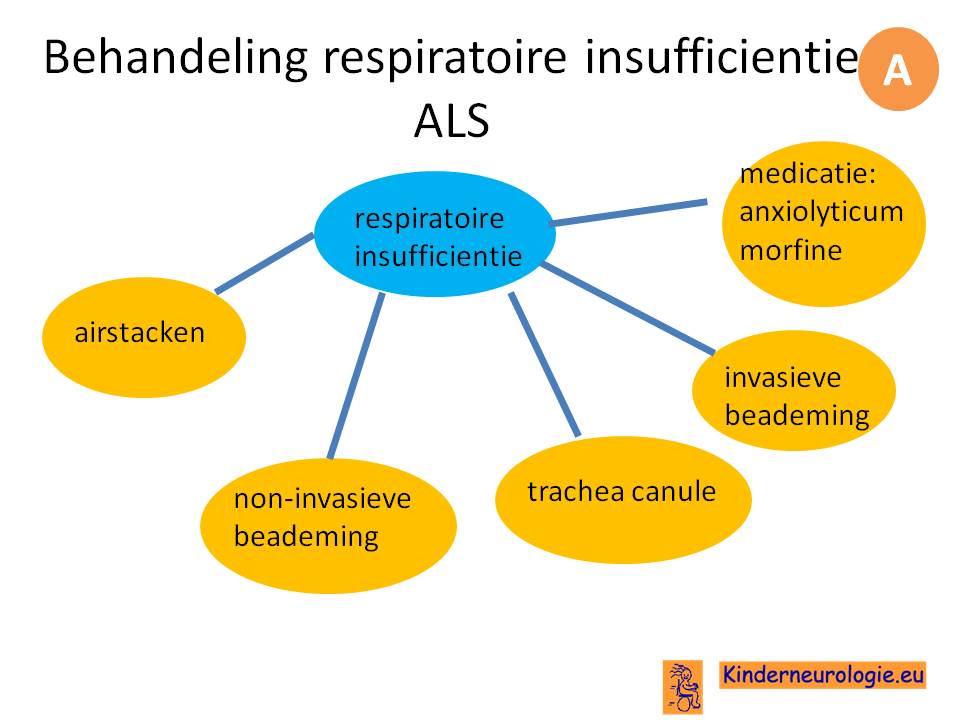

Er wordt onderzoek gedaan naar anti-sense oligonucleotide behandeling voor ALS.
Prognose
ALS is een progressieve aandoening.

SMA is een genetische aandoening die leidt tot progressief verlies van motore voorhoorncellen, leidend tot progressieve spierzwakte van de spieren van de armen en de benen. De spieren van het gezicht zijn gespaard bij kinderen met SMA.

SMA is een autosomaal recessieve aandoening. Er is sprake van een deletie op het SMN1-gen waardoor er minder survival-motor-neuron eiwit kan worden aangemaakt.

Op chromosoom 5 ligt ook een kopie van het SMN-gen, wat het SMN2-gen wordt genoemd. Met behulp van dit SMN2-gen kan nog wel een kleine hoeveelheid survival-motor-neuron eiwit aangemaakt worden. Vaak hebben kinderen met SMA meerdere kopieen van dit SMN2-gen. Een toename van de hoeveelheid kopieen van het SMN2-gen zorgt voor een milde beloop van de aandoening. Deze SMA-types hebben een hoger nummer gekregen.

De laatste jaren zijn er nieuwe behandelmogelijkheden gekomen om het tempo van toename van nieuwe klachten van SMA te vertragen. Het gaat om anti-sense oligonucleotide therapie (ASO-therapie) en gentherapie.
Bij gentherapie wordt door middel van een adeno-associated virus (AAV type 9: zolgensma) het ontbrekende gen in de motore voorhoorncel van een patient met SMA gebracht. Dit gen wordt niet geintegreerd in het eigen DNA, maar ligt los in de kern. Omdat motore voorhoorncellen niet meer delen is dit geen probleem. Aflezen van dit DNA zorgt voor de aanmaak van meer survival-motor-neuron eiwit waardoor het tempo van achteruitgang van de ziekte wordt afgeremd.
Sinds 2017 wordt deze behandeling in Nederland op onderzoeksbasis toegepast bij kinderen met SMA type 1. Deze kinderen bleken in staat te zijn om zelfstandig te leren zitten en hadden een grotere levensverwachting dan kinderen die niet behandeld zijn.
De tweede vorm van behandeling bij SMA is door middel van anti-sense oligonucleotide therapie (nursinersen). Door middel van dit gen wordt exon 7 van het SMN2-RNA wel afgelezen, waardoor een beter survival-motor-neuron eiwit wordt gemaakt met behulp van het SMN2-gen. ASO therapie passeert de bloedhersenbarriere niet, waardoor deze therapie intrathecaal moet worden gegeven.
De behandeling heeft het meest effect wanneer kinderen nog maar weinig of geen klachten hebben. Presymptomatische behandeling blijkt het grootste effect te hebben. Daarom komt SMA vanaf 2022 in de hielprikscreening.
Polio is een ziekte die zorgt voor schade aan de motore voorhoorncel. Door vaccinatie (DKTP-vaccin) komt deze ziekte niet meer voor in Nederland sinds 1993.

Er zijn in Nederland nog veel patienten die als kind polio hebben gehad. De helft van de patienten krijgt toename van klachten zonder dat hiervoor een goede verklaring te vinden is. Dit wordt het postpolio syndroom genoemd.



Links
www.spierziekten.nl
(Nederlandse vereniging voor mensen met een spierziekte)
Referenties
1. Biemond cursus neuromusculaire aandoeningen 2021
Laatst bijgewerkt: 17 december 2021 voorheen: 9 april 2016
Auteur: JH Schieving
Heeft uw kind nog andere symptomen, laat het ons weten.